观察和预处理用连字符连接代谢物和蛋白质/肽质谱数据集分析
这个例子展示了如何操作,从液相色谱进行预处理和可视化数据加上质谱(LC / MS)。这些大型和高维数据集是广泛用于蛋白质组学和代谢组学研究。可视化复杂肽或代谢物混合物提供了一个直观的方法来评估样本的质量。此外,有条不紊的校正和预处理可以导致自动化高通量分析样品允许准确识别重要的代谢产物在生物样品和特定的肽的特性。
介绍
在一个典型的带有连字符号的质谱实验、蛋白质和代谢物是从细胞,组织或体液,溶解在溶液中变性、酶消化成混合物。这些混合物分离通过高效液相色谱法(HPLC),毛细管电泳(CE),或气相色谱(GC)和耦合质谱仪鉴定方法,如电喷雾电离质谱(质),矩阵协助电离(MALDI或SELDI TOF-MS),或串联质谱分析(MS / MS)。
开放的数据存储库和mzXML文件格式
对于本例,我们使用一个测试样本LC-ESI-MS七蛋白质混合的数据集。这个示例中的数据(7 mix_std_110802_1)的生鱼片数据存储库。数据集分布与MATLAB®。完成这个例子中,您必须下载数据集到一个本地目录或你自己的库。或者,你可以尝试其他的数据集可以在其他公共数据库对蛋白质表达数据等肽图谱系统生物学研究所的[1]。
大多数当前的质谱仪可以使用mzXML翻译或保存采集数据模式。这个标准是基于XML(可扩展标记语言)的常见文件格式开发的生鱼片项目解决的挑战参与代表数据集来自不同制造商和从不同的实验设置到一个共同的和可扩展的模式。mzXML文件用连字符连接中使用质谱通常是非常大的。的MZXMLINFO函数允许您获取基本信息数据集没有读到内存中。例如,您可以检索的扫描,保留时间的范围,串联女士仪器(水平)的数量。
信息= mzxmlinfo (“7 mix_std_110802_1.mzxml”,“NUMOFLEVELS”,真正的)
信息=结构体字段:文件名:“7 mix_std_110802_1。mzXML FileModDate: 01 - 2月2013 11:54:30的文件大小:26789612 NumberOfScans: 7161开始时间:PT0.00683333S‘EndTime: PT200.036S”DataProcessingIntensityCutoff:“N / A”DataProcessingCentroided:“真正的”DataProcessingDeisotoped:“N / A”DataProcessingChargeDeconvoluted:“N / A”DataProcessingSpotIntegration:“N / A”NumberOfMSLevels: 2
的MZXMLREADMATLAB函数读取XML文档转换为一个结构。的字段扫描和指数被放置在第一级的输出结构改进对光谱数据的访问。剩下的mzXML根据解析文档树模式规范。此LC / MS数据集包含7161扫描与女士的两个层面。仅供本例中您将使用第一级扫描。二级光谱通常用于多肽/蛋白质识别,并在某些类型的后期工作流程分析。MZXMLREAD可以过滤所需的扫描没有加载所有数据到内存:
mzXML_struct = mzxmlread (“7 mix_std_110802_1.mzxml”,“水平”,1)
mzXML_struct =结构体字段:扫描:[2387×1 struct] mzXML: [1×1 struct]指数:[1×1 struct]
如果你收到任何错误相关的java堆内存或空间加载期间,尽可能增加java堆空间描述在这里。
质谱仪和实验条件有关的更详细的信息在地里被发现msRun。
mzXML_struct.mzXML.msRun
ans =结构体字段:scanCount: 7161开始时间:“PT0.00683333S”endTime:“PT200.036S”parentFile: [1×1 struct] msInstrument: [1×1 struct] dataProcessing: [1×1 struct]
为了便于数据的处理,MZXML2PEAKS函数提取的山峰从每个扫描成一个单元阵列(峰))和各自的保留时间为一个列向量(时间)。你可以提取光谱通过设置一定的水平水平输入参数。
(峰值、时间)= mzxml2peaks (mzXML_struct);numScans =元素个数(山峰)
numScans = 2387
的MSDOTPLOT函数创建一个概览显示最强烈的山峰在整个数据集。在这种情况下,我们只看到最强烈的离子强度的峰值5%通过设置输入参数分位数到0.95。
h = msdotplot(峰值、时间分位数的,.95);标题(“5%最强烈的山峰”)

还可以过滤峰值分别为每个扫描使用基本峰值强度的百分比。基峰是最强烈的峰发现在每个扫描[2]。这个参数自动给出大部分的光谱仪。这个操作需要查询到mxXML结构来获得基峰信息。注意,您也可以通过迭代找到基地峰值强度马克斯函数的峰值。
basePeakInt = [mzXML_struct.scan.basePeakIntensity] ';peaks_fil =细胞(numScans, 1);为i = 1: numScans h =山峰{我}(:,2)> (basePeakInt(我)。* 0.75);peaks_fil{我}={我}山峰(h:);结束谁(“basePeakInt”,“level_1”,“高峰”,“peaks_fil”)msdotplot (peaks_fil时间)标题(上面的山峰(0.75 x基地峰值强度)为每一个扫描的)
类属性名称大小字节basePeakInt 2387 x1 19096双峰值2387 x1 14031800细胞peaks_fil 2387 x1 289568细胞

您可以自定义过滤高峰使用的3 d概述STEM3函数。的STEM3需要把数据分成三个向量函数,其元素组成了三胞胎(保留时间、质量/电荷,强度值)表示每一杆。
peaks_3D =细胞(numScans, 1);为i = 1: numScans peaks_3D{我}(:[2 3])= peaks_fil {};我peaks_3D{}(: 1) =时间(我);结束peaks_3D = cell2mat (peaks_3D);图stem3 (peaks_3D (: 1), peaks_3D (:, 2), peaks_3D (:, 3),“标记”,“没有”)轴([0 12000 400 1500 0 1 e9])视图(60 60)包含(的保留时间(秒))ylabel (“质量/电荷(M / Z)”)zlabel (“相对离子强度”)标题(上面的山峰(0.75 x基地峰值强度)为每一个扫描的)

你可以画出彩色的使用补丁函数。每三个一组peaks_3D,交错一个新的三联体的强度值设置为零。然后创建一个颜色向量依赖杆的强度。一个对数变换增强colormap的动态范围。交叉三胞胎分配一个南,所以补丁函数不画线连接相邻的茎。
peaks_patch = sortrows (repmat (peaks_3D 2 1));peaks_patch(2:2:结束,3)= 0;col_vec =日志(peaks_patch (:, 3));col_vec(2:2:结束)=南;图块(peaks_patch (: 1), peaks_patch (:, 2), peaks_patch (:, 3), col_vec,…“edgeColor”,“平”,“markeredgecolor”,“平”,“标记”,“x”,“MarkerSize”3);轴([0 12000 400 1500 0 1 e9])视图(90、90)包含(的保留时间(秒))ylabel (“质量/电荷(M / Z)”)zlabel (“相对离子强度”)标题(上面的山峰(0.75 x基地峰值强度)为每一个扫描的)

视图(40、40)

创建热图的LC / MS数据集
业内普遍技术处理峰值信息(又名质心数据)而不是原始信号。这可能节省内存,但一些重要的细节是不可见的,特别是当它是必要的检查与复杂混合物样品。进一步分析这个数据集,我们可以创建一个共同的网格的质量和电荷密度。因为不是所有的扫描有足够的信息来重建原始信号,我们使用一个峰值保持重采样方法。通过选择适当的参数MSPPRESAMPLE功能,可以确保不会丢失光谱的分辨率,和的最大值峰相关原始信息达到顶峰。
(MZ, Y) = msppresample(峰值,5000);谁(“MZ”,“Y”)
类属性名称大小字节MZ 5000 x1 20000单Y 5000 x2387 47740000单
这个矩阵的离子强度,Y,您可以创建一个彩色的热图。的MSHEATMAP函数自动调整colorbar用来显示显著峰值与热与冷颜色。颜色和吵闹的山峰基于聚类的算法是重要的山峰和噪声峰值的估算与采用混合高斯模型的方法。此外,您可以使用中点输入参数选择任意阈值分离噪声峰值与重要的山峰,或者你可以交互式地colormap转移到隐藏或显示峰值。在处理热点图时,通常显示的对数离子强度,提高colormap的动态范围。
fh1 = msheatmap (MZ,时间日志(Y),“决议”、1。“范围”900年[500]);标题(“原来的LC / MS数据集”)

可以放大到小区域感兴趣的观察数据,交互地或编程方式使用轴函数。我们观察到一些地区相对高离子强度。这些代表多肽在生物样本。
轴([527 539 385 496)

您可以覆盖原始的LC / MS的峰值信息数据集。这让你评估的性能peak-preserving重采样操作。您可以使用返回的句柄,隐藏/显示点。
dp1 = msdotplot(峰值、时间);

校准的质量/电荷位置峰值常见的网格
二维峰似乎吵或他们不显示一个紧凑的形状在连续的光谱。这是一个常见的问题对于许多质谱仪。质量/电荷值的随机波动从山峰中提取的复制概要文件报告的范围从0.1%到0.3% [3]。这样的变化可以由几个因素引起的,例如探测器校准差,低信噪比、峰值提取算法的问题。的MSPALIGN函数实现了先进的数据装箱算法,同步所有的光谱数据设置为一个共同的质量/电荷网格(CMZ)。CMZ可以选择任意或可以估计在分析数据(2、4、5)。峰值匹配过程可以使用最近邻规则或动态编程对齐。
(CMZ peaks_CMZ] = mspalign(峰);
重复的可视化过程一致的山峰:执行峰值保持重采样,创建一个热图,覆盖对齐峰信息,放大同一地区感兴趣的。怎样的光谱时,可以区分的一些肽的同位素模式。
[MZ_A, Y_A] = msppresample (peaks_CMZ, 5000);fh2中= msheatmap (MZ_A、时间日志(Y_A),“决议”10,“范围”900年[500]);标题(的LC / MS数据集的质量/电荷校准CMZ”)dp2 = msdotplot (peaks_CMZ、时间);轴([527 539 385 496)

在本地校准的质量/电荷位置峰值
MSPALIGN计算单个CMZ整个LC / MS数据集。这可能不是理想的情况与更复杂的多肽混合物样品和/或代谢产物比在这个例子中所用的数据集。对于复杂的混合物,可以使每个光谱本地组只包含信息的光谱峰保留时间(高强度的)相似,否则校准过程小山峰地区(低密度)可以被其他山峰有偏见的质量/电荷值相似,但不同的保留时间。进行精细标定,可以采用SAMPLEALIGN函数。这个函数是一个泛化的约束动态时间扭曲(CDTW)算法通常用于语音处理[6]。在接下来的为循环,我们保持缓冲强度之前的光谱(LAI)保持一致。光谱的离子强度是按比例缩小的匿名函数科幻小说(在SAMPLEALIGN),以减少大峰之间的距离。SAMPLEALIGN降低了整体所有匹配点的距离和在必要时引入了差距。在本例中,我们使用一个更精细的MZ向量(FMZ),这样我们保持正确的值的质量/电荷山峰尽可能多。注意:这可能需要一些时间,因为CDTW算法执行2387次。
科幻小说= @ (x) 1-exp (x / 5 e7);%尺度函数DF = @ (R, S)√(科幻小说(R(:, 2))科幻(S (:, 2)))。^ 2 + (R (: 1) - s (: 1)) ^ 2);FMZ = (500:0.15:900) ';%设置一个细MZ向量赖= 0(大小(FMZ));%初始化缓冲一致性强度peaks_FMZ =细胞(numScans, 1);为我= 1:numScans%显示进展如果~ rem(我250)流(“% d…”,我);结束%对齐山峰在当前扫描赖[k] = samplealign ([FMZ,赖),双(峰值{我}),“乐队”,1.5,“差距”(0,2),“距离”DF);%更新赖缓冲区赖赖= *为;赖赖(k) = (k) +山峰{我}(j, 2);%保存校准peaks_FMZ{我}= [FMZ (k)的峰值{我}(j, 2)];结束
250年……500年……750年……1000年……1250年……1500年……1750年……2000年……2250年……
重复的可视化过程并缩放到感兴趣的地区。
[MZ_B, Y_B] = msppresample (peaks_FMZ, 4000);fh3 = msheatmap (MZ_B、时间日志(Y_B),“决议”10,“范围”900年[500]);标题(的LC / MS数据集质量/电荷校准本地的)dp3 = msdotplot (peaks_FMZ、时间);轴([527 539 385 496)

作为最后一步改善图像,你可以在色谱方向应用高斯滤波器平滑整个数据集。
Gpulse = exp(约*(10:10)。^ 2)。/笔(exp(约* (10:10)。^ 2));YF = convn (Y_B Gpulse,“相同”);fh4 = msheatmap (MZ_B、时间日志(YF),“决议”10,“限制”900年[500]);标题(最终提高LC / MS数据集的)dp4 = msdotplot (peaks_FMZ、时间);轴([527 539 385 496)

你可以链接两个热点图的坐标轴,交互地或编程方式比较两个数据集之间的区域。在这种情况下,我们比较原始和最终提高LC / MS矩阵。
linkaxes (findobj ([fh1 fh4),“标签”,“MSHeatMap”)轴([521 538 266 617)


使用的总离子色谱提取光谱
一旦LC / MS数据集是平滑并重新取样到常规的网格,可以提取信息最丰富的光谱通过观察的局部极大值的总离子色谱图(TIC)。抽搐是直接计算加法YF的行。然后,使用MSPEAKS函数来找到的保留时间值提取选定的光谱的子集。
抽搐=意味着(YF);pt = mspeaks(时间、抽搐的“乘数”10“overseg”,100,“showplot”,真正的);标题(的山峰中提取的总离子色谱图(TIC) ')pt (pt(: 1) > 4000年:)= [];%去除光谱超过4000秒numPeaks =大小(pt, 1)
numPeaks = 22

创建一个3 d的情节所选的光谱。
xRows = samplealign (pt (: 1),“宽度”1);%发现指数每一个峰值的时间xSpec = YF (:, xRows);%得到信号的情节图;持有在盒子在plot3 (repmat (MZ_B 1 numPeaks) repmat (1: numPeaks,元素个数(MZ_B), 1), xSpec)视图(85)ax = gca;斧子。YTick = 1:numPeaks; ax.YTickLabel = num2str(time(xRows)); axis([500 900 0 numPeaks 0 1e8]) xlabel(“质量/电荷(M / Z)”)ylabel (“时间”)zlabel (“相对离子强度”)标题(“提取光谱子集”)

覆盖标记提取的光谱的增强的热图。
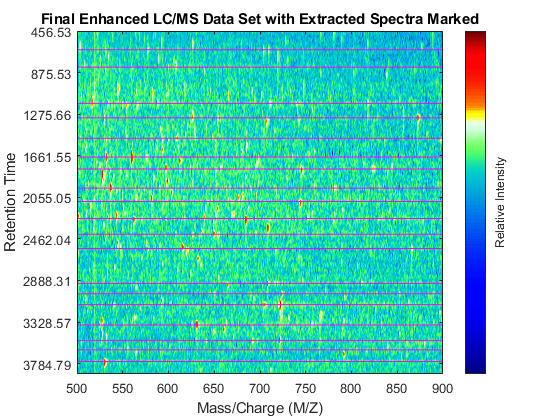
linkaxes (findobj (fh4,“标签”,“MSHeatMap”),“关闭”)图(fh4)在为i = 1: numPeaks情节(1500 [400],xRows([我]),“米”)结束轴dp4 ([500 900 100 925])。可见=“关闭”;标题(的最终提高LC / MS数据集提取光谱标志的)

引用
[1]Desiere, f . et al .,“肽图谱项目”,核酸研究,34:D655-8, 2006。
[2]Purvine, S。Kolker, N。,和Kolker, E., "Spectral Quality Assessment for High-Throughput Tandem Mass Spectrometry Proteomics", OMICS: A Journal of Integrative Biology, 8(3):255-65, 2004.
[3]伤势严重,响亮的,et al., "Alignment of high resolution mass spectra: Development of a heuristic approach for metabolomics", Metabolomics, 2(2):75-83, 2006.
[4]Jeffries, N。,"Algorithms for alignment of mass spectrometry proteomic data", Bioinformatics, 21(14):3066-3073, 2005.
[5],W。,et al., "Multiple peak alignment in sequential data analysis: A scale-space based approach", IEEE®/ACM Trans. Computational Biology and Bioinformatics, 3(3):208-219, 2006.
[6]Sakoe、h和千叶。,"Dynamic programming algorithm optimization for spoken word recognition", IEEE Trans. Acoustics, Speech and Signal Processing, ASSP-26(1):43-9, 1978.