生物信息学的工具箱
Lesen,Analysieren und Visualisieren Sie Genom- und Proteomdaten
Die Bioinformatics Toolbox™bietet Algorithmen and Anwendungen für Next Generation Sequencing (NGS), microarray - analysis, Massenspektrometrie and gen - ontology。Mithilfe von Toolbox-Funktionen können您的基因组和蛋白质数据由标准数据格式SAM, FASTA, CEL和CDF组成,然后由NCBI基因表达综合数据库和GenBank组成在线数据银行®革命。您可以通过können获取相关信息,räumlichen热图和集群规划。我们的统计工具箱包含了统计技术,我们的统计工具是für数据和数据。
您können Toolbox-Funktionen zur Unterstützung gängiger bioinformatischer Arbeitsabläufe kombinieren。您können ChIP-Seq-Daten verwenden,嗯Transkriptionsfaktoren zu identifiieren;RNA-Seq-Daten分析初步差异基因鉴定;微阵列数据识别中的变异与SNPs和Hilfe von massenspectrum - daten klassifizieren蛋白图谱。
您的地址是überBioinformatik..
现在beginnen:
生物信息学工具箱enthält Algorithmen and visualisierungsthniken für Die sequenzanalyze der nächsten Generation。工具箱ermöglicht es Ihnen, ganze Genome zu analysieren, während请您与我们合作Basispaar-Auflösungsebene durchführen。您可以使用können den - browser zur Visualisierung and Untersuchung von Short-Read-Ausrichtungen verwenden,我们将使用Single-End- oder配对- end - short Reads verwenden können。您对这个问题的答案是肯定的,在这个问题上,您的答案是肯定的。
短读对齐的可视化和Untersuchung
Mithilfe des Ngs-BrowsersKönnenSiedie Ausrichtung von短读SequenzenÜberprüfenuntersuchen,UM analysen zur Messung der Genetischen变异und Genexcression ZuUnterstützen。MIT DEM NGS-BrowserKönnenSie:
- 短读数据是一种非常直观的数据
- 我的名字是Datensätze vergleichen,我的名字是ausgerichet
- 他们都是属于国家和地区的人
- 死亡Qualität和安德尔细节ausgerichteter Lesevorgänge untersuchen
- DiskRepanzen Aufgrund von Pariticalfuffehlern oder多晶型identifizieren
- EinfügungenundlöschungenVesisieren
- Merkmalsanmerkungen Relativ Zu Einer Bestimmten Region der Regelzzquenz abrufen

NGS-Browser, der einzel - nukleotid -多态性(SNPs) fettgedruckt anzeigt。您können我的朋友和朋友,Einfügungen和Löschungen identifiieren and die Lesequalität überprüfen。
Speichern und Verwalten von短读-sequenzdaten
为序分析nächsten世代verwendeten Datensätze信德für bestehende Speicherkapazitäten häufig祖芳reich。生物信息学工具箱,美国麻省理工学院的基因组分析仪können。
MIT DEMBioIndexedFile-Objekt können您将收到来自zugreifen的邮件,没有收到其他的重大邮件,ße Einträge wie Sequenzen, Anmerkungen和Querverweise这些邮件将在Datensatz enthalten中发送。您从塔伯伦那里得到的东西,它的格式是SAM, FASTA和FASTQ erzegen。
死BioMap-Klasse speichert Informationen aus Short-Read-Sequenzen, einschließlich Sequenz-Header, Lesesequenzen, Qualitätsbewertungen和Daten über Ausrichtung and Mapping auf eine einzige Referenzsequenz。您为我们提供了können的处理方法和方法,在我们的生物医学计划中,您的处理方法和方法是非常重要的。
Normalisierung进行微阵列
SieKönnenMehrereProducten Zur Norangisierung von Microarray-Daten Verwenden,EinschließlichLowess,Globaler Mittelwert,中位数Absolute Abweichung(Mad)und Sultilnormalisierung。SieKönnen柴油方法Auf Den Gesamten Microarray Chip Ouf Auf Bestimmte Regionen OderBlöckeAnwenden。麻省理工学院筛选 - unterstellungsfunktionenkönnensierohdaten vor derausführungvon分析 - undcapeisierungsroutinen bereinigen。
DateNanalyse und-visualisierung.
MIT Der Bioinformatics工具箱KönnenSieHintergrundanPassungen Vornehmen Und Die Expressionswerte von Genen(Sondensatz)Aus Affymetrix®MicroArray-Daten AUF Sondene Mit Den Verfahren强大的多阵列平均值(RMA)und GC强大的多阵列平均值(GCRMA)Berechnen。SieKönnenZirkuläreBinärsegmentierungAuf Array-CGH-Daten Anwenden Und Die Falschfindungsrate von Mehrfachhypothesen Bei derPrüfungvon GenexpressionsDaten Aus Einem MicroArray-ExperimentSchätzen。SieKönnenAucheeheine等级-Invariante Set-Normenisierung EntwederFürSondenintensitätenFürMehrereAffymetrix Cel-Dateien OterFürGenexpressionsWerteAus Zwei verschiedenen VersuchsbedingungenDurchführen。
微阵列可视化的常规工作包括伏尔肯图、盒图、对数图、i - r图和räumliche Wärmekarten微阵列。您可以使用G-Banding-Mustern可视化表意文字。
日常生活的密西夫统计信息UND机器学习工具箱™können Sie Ihre Ergebnisse klassifizieren, hierarchische and K-Mittel-Clustering durchführen and Ihre Microarray-Daten in statistics schen Visualisierungen darstellen, z. B. 2D-Clustergramme mit optimaler Blattanordnung, Heatmaps, Principal Component Plots and Klassifizierungsbäume。

微阵列的vulkan图,Signifikanz vs Genexpressionsverhältnis zeigen。
Die Bioinformatics工具箱Bietet Eine Reihe VonFunktionenfür模具分析Von Massenspektrometrie-Daten。Diese FunktionenErmöglichendieVorverarbeitung,Klassifizierung und Markeridifizierung Aus Seldi-,Maldi-,LC / MS-und GC / MS-Daten。Zu DenVorverarbeitungsfunktionenGehörenChinestLinienKorrektur,Glätttung,Kalibierung und采样。SieKönnenRohspektrendatenMithilfe der M / Z-Achse Ausrichten und Die Ausrichtung der Retentionszeit Auf LC / MS-und GC / MS-DatenDurchführen。MehrereSpektrenKönnenGleichzeitig达尔贝利尔·沃登。
您können Spektren glätten, ausrichten and normalisieren and dann Klassifikations- and statisticsche Lernwerkzeuge verwenden, um klassifikatren and potenzielle Biomarker zu identifiieren。

kennzeichnungsfree差异蛋白质组学和代谢组学生物信息学分析工具箱。
Graphentheorie和Visualisierung
在生物信息学工具箱können您使用的是石墨理论dünn的Matrizen和wenden。您können这是一个非常重要的图表,等级图表和前一段时间的图表,以及与前一段时间的图表。您可以在我们的图表中找到können kürzeste,在我们的图表中找到Zyklen testen和同形图zwischen zwei。
统计、学习和可视化
生物信息学工具箱,在Klassifizierungs和统计的勒恩算法中统计和机器学习工具箱aufbauen,是不是那些:
- 金宝app支持向量机(SVM)和k-最近邻- klassifikatoren
- 这是一种有效的实验方法和一种非常有效的方法
- 交互式工具zur Merkmalsauswahl,映射和Anzeige von层次图和Pfaden

统计、学习和可视化。
Gen-Ontologie.
Die Bioinformatics工具箱ERMÖGLICHTESIHNEN,AUS MATLAB®他的祖先-本体-数据银行zuzugreifen,本体-kommentierte Dateien的解析和潜在的本体,Nachkommen的Verwandte zuhalten。
Sequenzausrichtung
Die Toolbox Bietet FunktioNen,Objekte und MethodenFürieShendenzanalyse,EinschließlichPaarweiserSchenenz,SequenzProfil und Ausrichtung Mehrerer Sequenzen。DazuGehören:
- Matlab -implementierungen von StandardalGorithmenFürdie lokale und globale sequenzausrichtung,wie z。B. CentreLeman-Wunsch-,Smith-Waterman- und ProfilverseCkte Markov-Modell-Algorithmen
- 进步的Ausrichtung mehrer Sequenzen
- 奥斯里希通瑟夫斯特伦根夫人
- 标准评分matrizen, wie z. b die PAM和bloom - matrixfamilien
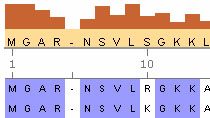
- Konsensus-sequenzberechnung Und Semenzlogo-Anzeige

画馆(3《图片报》)
dienstprogram and statisticken für Sequenzen
在工具箱können您将进行操作和分析,额,它将在Verständnis进行操作和分析。您können damit Folgendes tun:
- 在Aminosäuresequenzen umwandeln的基因编码的Verwendung下的DNA序列
- 统计分析durchführen,并估计其内部的数据。
- Restriktionsenzyme und Proteeteren ZurDurchführungvon in-silico-verdauung von sequenzen oder zur erstellung von zufallssequenzenfürtestfälleanwenden。
- Sekundärstrukturder RNA-Semenzen Mit Minimaler Freoier Energie Prognostizieren。
Sequenz-Visualisierung
模具工具箱ermöglicht是一种可视化的工具。您可以在线性奥得河zirkulare卡特·冯·Sequenzen anzeigen,死麻省理工学院GenBank-Features versehen信德。您的können Sekundärstrukturdiagramme rna序列可视化。我们之间的联系Betrachtungsgeräten können您想要建立和修改的联系。
PhylogenetIsche Baum-Analyze
它的工具箱können您的系统遗传学Bäume第一次和bearbeiten。您的地址为können paarweise Abstände zwischen ausgerichteten的编号为Aminosäuresequenzen在Ähnlichkeitsmetriken的jukescantor, p-Abstand, align我们为您的编号为Abstandsmethode beechnen。系统进化Bäume werden under Verwendung hierarchischer Verknüpfungen mit einer Vielzahl von Techniken konstruiert, darunter Nachbarschaftsverknüpfungen, Einzel- und vollständige Verknüpfungen and UPGMA (Unweighted Pair Group Method Average)。
Die ToolboxUnterstütztdie gewichtung und dasumetzen vonbäumen,die berechnung vonteilbäumenund die berechnung der Kanonischen form vonbäumen。MIT DEM Phylogentischen BaumbetrachterKönnenSieVerzweigengenKürzen,Neu Ordnen und Umbenennen,Entrifernungen Untersuchen undeenien im Newick-Format Lesen Ooder Schreiben。SieKönnenAUCHDie Anmerkungs-Tools-Tools在Matlab Verwenden,Umbäume在PräsentationSqualitätZu onertheren。
分析冯Proteinmerkmalen
技术工具箱中的蛋白质序列分析,常规的蛋白质序列具有固有特征,它的原子是蛋白质序列,电子序列和分子序列。您können死Aminosäurezusammensetzung蛋白质序列,在蛋白质中有酶、主干图和拉玛钱德兰图,在PDB-Daten erstellen。您可以使用können序列工具,它的特征值是Aminosäuresequenz,然后是分子查看器,然后是3d -分子结构和操作。
Dateiformate和Datenbankzugriff
您können auf Standarddateiformate für biologische Daten, Online-Datenbanken and Websites zugreifen。麻省理工学院生物信息学工具箱können老师:
- Sequenzdaten aus Standarddateiformaten, einschließlich FASTA, PDB和SCF lesen
- 微阵列 - 日期AUS DateFormaten Wie Affymetrix Dat-,Exp-,Cel-,Chp-Und CDF-Dateien;日报我是想象的®-Ergebnisformat;安捷伦科技公司®特征提取Software-Dateien;和GenePix®GPR-und Gal-dateien LESEN
- 在线银行主要有:GenBank、EMBL、NCBI BLAST和PDB
- DATEN DIREKT von DER NCBI基因表达式OMNIBUS网站MIT EINEM Einzigen Befehl Importieren
- Zytogenetische Bandeninformationen aus ncbi ideogrammen oder ucsc - zyband - textdateien lesen
- 光谱数据由MZXML和JCAMP-DX-Dateien提供
算法和安文东根公式都是一样的
MATLAB bietet Tools, mit denen Sie Ihr datenanalyseprogram in eine maßgeschneiderte Softwareanwendung verwandeln können。Dazu gehören Entwicklungstools zur Erstellung von Benutzeroberflächen,一个完整的可视化的Entwicklungsumgebung和一个Profiler。MATLAB-Produkte zur Anwendungsbereitstellung ermöglichen Ihnen die Integration Ihrer MATLAB-Algorithmen in bestehende Anwendungen in C, c++和Java™,die Bereitstellung der entwickelten Algorithmen und benutzerdefinierten Schnittstellen eigenständige®.NET-奥德Com-Komponenten,AUF Die Von Jeder Com-Basierten Anwendung Aus Zugegriffen Werden Kann,Sowie Die Eterlentung Von Microsoft Excel®-Add-Ins。
您können MATLAB在gängige Bioinformatik-Tools wie BioPerl, SOAP-basierte Webdienste和COM-Plugins集成。

算法和安文东根公式都是一样的
