使用全基因组数据
这个例子展示了如何为序列数据创建一个内存映射文件,并在不将所有基因组序列加载到内存的情况下使用它。人类、小鼠、大鼠、河豚和其他几种模式生物的全基因组都是可用的。对于许多这样的生物来说,一条染色体可以有几亿对碱基对长。处理如此大的数据集可能具有挑战性,因为您可能会遇到所使用的硬件和软件的限制。这个例子展示了在MATLAB®中绕过这些限制的一种方法。
大数据集处理问题
解决需要处理和分析大量数据的技术计算问题对计算机系统提出了很高的要求。大型数据集在处理过程中占用大量内存,并且可能需要许多操作来计算一个解决方案。从大型数据文件中访问信息也需要很长时间。
然而,计算机系统有有限的内存和有限的CPU速度。可用资源因处理器和操作系统而异,后者也会消耗资源。例如:
32位处理器和操作系统可以寻址到2^32 = 4,294,967,296 = 4gb的内存(也称为虚拟地址空间)。Windows XP和Windows 2000只为每个进程(如MATLAB)分配2 GB的虚拟内存。在UNIX®上,分配给进程的虚拟内存是系统可配置的,通常在3gb左右。执行计算的应用程序,如MATLAB,除了用户任务外还需要存储。处理大量数据时的主要问题是,程序的内存需求可能超过平台上的可用内存。例如,在Windows XP上,当数据需求超过约1.7 GB时,MATLAB会产生“内存不足”错误。
有关内存管理和大数据集的详细信息,请参见性能和内存.
在典型的32位机器上,您在MATLAB中可以处理的单个数据集的最大大小是几百MB,或者大约是一个大染色体的大小。文件的内存映射允许MATLAB绕过这个限制,使您能够以一种直观的方式处理非常大的数据集。
全基因组数据集
最新的全基因组数据集可从运用网站.数据以几种格式提供。当新的序列信息可用时,这些信息会定期更新。本例将使用以FASTA格式存储的人类DNA数据。染色体1(在2009年9月发布的GRCh37.56中)是一个65.6 MB的压缩文件。解压后的文件大小约为250MB。MATLAB每个字符使用2个字节,因此如果将文件读入MATLAB,它将需要大约500MB的内存。
本示例假设您已经下载了FASTA文件并将其解压缩到本地目录中。更改变量的名称FASTAfilename如果合适。
FASTAfilename =“Homo_sapiens.GRCh37.56.dna.chromosome.1.fa”;fileInfo = dir(which(FASTAfilename))
fileInfo =带有字段的struct: name: ' homo_sapiens . grch37 .56.dna.染色体。fa' folder: '/mathworks/hub/qe/test_data/Bioinformatics_Toolbox/v000/demoData/biomemorymapdemo' date: '01-Feb-2013 11:54:41' bytes: 253404851 isdir: 0 datenum: 7.3527e+05
内存映射文件
内存映射允许MATLAB访问文件中的数据,就像它在内存中一样。您可以使用标准的MATLAB索引操作来访问数据。请参阅相关文档memmapfile了解更多细节。
您可以映射FASTA文件并直接从那里访问数据。但是FASTA格式文件包含新的行字符。的memmapfile函数以与所有其他字符相同的方式处理这些字符。在内存映射文件之前删除这些将使索引操作更简单。此外,内存映射不能直接与字符数据工作,所以你必须将数据作为8位整数(uint8类)。这个函数nt2int在生物信息学工具箱™中可用于将字符信息转换为整数值。int2nt用于转换回字符。
首先打开FASTA文件并解压缩头文件。
fidIn = fopen(FASTAfilename,“r”);header = fgetl(fidIn)
header = '>1 dna:染色体染色体:GRCh37:1:1:249250621:1'
打开要被内存映射的文件。
[fullPath, filename, extension] = fileparts(FASTAfilename);mmFilename = [filename .“功能”fidOut = fopen(mmFilename,' w ');
mmFilename = ' homo_sapiens . grch37 .56.dna.染色体1.mm'
以1MB的块读取FASTA文件,删除新的行字符,转换为uint8,并写入MM文件。
newLine = sprintf(' \ n ');blockSize = 2^20;而~ feof (fidIn)%读取数据charData = fread(fidIn,blockSize,“*字符”)”;%删除新行charData = strrep(charData,newLine,”);%转换为整数intData = nt2int(charData);写入新文件写入文件(fidOut intData,“uint8”);结束
关闭文件。
文件关闭(fidIn);文件关闭(fidOut);
新文件与旧文件大小相同,但不包含新行或头信息。
mmfileInfo = dir(mmFilename)
“hom_sapiens . grch37 .56.dna.染色体1.”/tmp/Bdoc22a_1891349_66459/tp4e06d876/ex57563178' date: '26-Feb-2022 12:38:14' bytes: 249250621 isdir: 0 datenum: 7.3858e+05
访问内存映射文件中的数据
命令memmapfile构造一个memmapfile对象,该对象将新文件映射到内存。为了做到这一点,它需要知道文件的格式。这个文件的格式很简单,但是可以映射更复杂的格式。
chr1 = memmapfile(mmFilename,“格式”,“uint8”)
chr1 = Filename: '/tmp/Bdoc22a_1891349_66459/tp4e06d876/ex57563178/ homo_sapiens . grch37 .56.dna.染色体。'可写:false Offset: 0格式:'uint8'重复:Inf Data: 249250621x1 uint8 array
MEMMAPFILE对象
memmapfile对象具有各种属性。文件名存储文件的完整路径。可写的指示数据是否可以修改。注意,如果您修改了数据,这也将修改原始文件。抵消允许指定任何标头信息所使用的空间。格式数据格式。重复用于指定多少块(如由格式)以绘制地图。这对于限制用于创建内存映射的内存数量非常有用。可以以与其他MATLAB数据相同的方式访问这些属性。更多详细信息请参见type帮助memmapfile或医生memmapfile.
chr1.Data (1:10)
Ans = 10x1 uint8列向量15 15 15 15 15 15 15 15 15 15 15 15 15 15
您可以使用索引操作访问数据的任何区域。
chr1.Data (10000000:10000010) '
Ans = 1x11 uint8行向量11 2 2 2 2 2 3 4 2 4 2 2 2
记住,核苷酸信息被转换为整数。你可以用int2nt来获取序列信息。
int2nt (chr1.Data(10000000:10000010)”)
ans = 'AACCCCGTCTC'
或使用seqdisp显示序列。
seqdisp (chr1.Data(10000000:10001000)”)
ans = 17x71字符数组' 1 AACCCCGTCT CTACAATAAA TTAAAATATT AGCTGGGCAT GGTGGTGTGT GCTTGTAGTC' ' 61 CCAGCTACTT ggcgggtgaa TCATCCAAGC CTTGGAGGCA gaggttttgcag ' ' 121 TGAGCTGAGA TTGTGACACT gttggagagc cttggaggcag ' ' 181 AAAAAACAAA aaacaaaaaaaaaa CAAACCACAA AACTTTCCAG gttggaggcag ' ' 181 AAAAAACAAA aaacaaa aaacaaaa CAAACCACAA AACTTTCCAG GTAACTTATT aaaacgtt ' ' 241 TTTTGTTTGT tttgtgg tttgtgg AGTGGAGCAA' ' 301 tctcagcca ctgcactc CGCCTCCCGG gttccccccc ' ' 361 gagtagtaggactaggc ACCCGCCACCAcgcccagct tatttttttt gtatttttta ' ' 421 gtagagacgg ggtttcatcg tgttccag gatttttccag atctccttgac ctcgtgcc ' ' 481 gcccacctca gcctcccaaa gtgctgggat tacagcgtg agccactgca cccggcctag ' ' 541 tttttgtata ttttttttag tagagaccagg gtttcaccat gttagccccaag atggcgtga ' ' 601 tcttgacc tcgtgatccg CCCGCCTCGG cctcccaag tgctcgtga ' ' 661 gccaccgcac acagcattaa atttttttt atttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt atttttttttt attattattata ' ' 781 taatgacaaa aaattactac ' ' 781 taatgacaaa aaattactac' 841 atgatatgca aacataaaca agtattatac ccagaagtgt aatttattgt agtacatct ' ' 901 tatgtataat agtttagtgg atttttcctg gaaattgtcc attttaattttttaag ' ' 961 tctgtggat tttccagtaa aagtcaaggc aaacccaaga t ' '
全染色体分析
既然可以很容易地访问整个染色体,就可以分析数据了。这个例子展示了一种查看沿着染色体的GC内容的方法。
提取500000bp的块并计算GC内容。
计算要使用多少块。
numNT = numel(chr1.Data);blockSize = 500000;numBlocks = floor(numNT/blockSize);
在处理大型数据集时,提高MATLAB性能的一种方法是为数据“预先分配”空间。这使得MATLAB可以为所有数据分配足够的空间,而不必将数组扩展成小块。这将加快速度,还可以防止数据过大而无法存储的问题。有关预分配数组的详细信息,请参见://www.tatmou.com/matlabcentral/answers/99124-how-do-i-pre-allocate-memory-when-using-matlab
方法预分配数组的一种简单方法是使用0函数。
ratio = 0 (numBlocks+1,1);
循环遍历数据寻找C或G,然后将这个数字除以A、T、C和G的总数。这将花费大约一分钟的时间。
A = nt2int(“一个”);C = nt2int(“C”);G = nt2int(‘G’);T = nt2int(“T”);为count = 1:numBlocks%计算块的索引start = 1 + blockSize*(count-1);stop = blockSize*count;%提取区块block = chr1.Data(start:stop);%找到GC和AT内容gc = (sum(block == G | block == C));at = (sum(block == A | block == T));%计算GC与已知核苷酸总数的比率比率(计数)= gc/(gc+at);结束
最后一个块比较小,所以把它当作一个特例。
block = chr1.Data(stop+1:end);gc = (sum(block == G | block == C));at = (sum(block == A | block == T));Ratio (end) = gc/(gc+at);
智人1号染色体GC含量图
xAxis = [1:blockSize:numBlocks*blockSize, numNT];情节(xAxis比率)包含(“碱基对”);ylabel (“相对GC内容”);标题(“人类1号染色体的相对GC含量”)
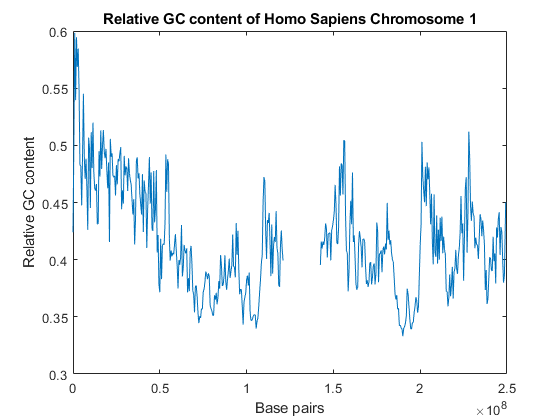
地块中心约140Mbp的区域是Ns的大片区域。
seqdisp (chr1.Data (140000000:140001000))
ans = 17 x71 char数组' 1 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“61年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“121年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“181年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“241年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“301年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN”“361年NNNNNNNNNN NNNNNNNNNN NNNNNNNNNNNNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“421 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“481 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“541 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“601 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“661 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“721 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“781 NNNNNNNNNN NNNNNNNNNNNNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“841 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“901 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN“961 NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN NNNNNNNNNN N '
寻找GC含量高的区域
你可以用找到识别GC含量高的区域。
指数= find(ratio>0.5);range = [(1 + blockSize*(indexes -1)), blockSize* indexes];流('区域%d:%d有GC内容%f\n',范围,比率(指数)”)
区域500001:1000000 GC含量0.501412区域1000001:1500000 GC含量0.598332区域1500001:2000000 GC含量0.539498区域2000001:2500000 GC含量0.594508区域2500001:3000000 GC含量0.568620区域3000001:3500000 GC含量0.584572区域3500001:4000000 GC含量0.548137区域6000001:6500000 GC含量0.545072区域9000001:9500000 GC含量0.506692区域10500001:11000000 11500001:12000000 GC含量0.511386区域GC含量0.519874地区16000001:16500000有GC含量0.513082地区17500001:18000000有GC含量0.513392地区215000001:22000000有GC含量0.505598地区156000001:156500000有GC含量0.504446地区156500001:157000000有GC含量0.504090地区201000001:201500000有GC含量0.502976地区228000001:228500000有GC含量0.511946
如果要删除临时文件,必须先清除memmapfile对象。
清晰的chr1删除(mmFilename)
您也可以从以下列表中选择网站: