微分分析复杂的蛋白质和代谢物混合物使用液相色谱/质谱(LC / MS)
这个例子显示了如何SAMPLEALIGN函数可以正确的非线性弯曲用连字符连接质谱色谱维度的数据集不需要样品的完整识别化合物和/或内部标准的使用。这样的纠正扭曲的一对(或一组)之间血缘关系样本,微分分析增强,可以自动化。
介绍
使用复杂的多肽和代谢物在LC / MS混合物需要label-free校准程序。这种类型的数据的分析需要寻找生物统计学意义差异相关的数据集,而不需要一个完整的标识样本中所有的化合物(多肽/蛋白质或代谢物)。在两个维度比较化合物需要对齐,mass-charge维度和保留时间维度[1]。例子中原始质谱数据预处理和观察和预处理用连字符连接代谢物和蛋白质/肽质谱数据集分析,你可以学习如何使用MSALIGN,MSPALIGN,SAMPLEALIGN函数经校准的不同类型的异常或质量/电荷维度。在本例中,您将学习如何使用SAMPLEALIGN函数也正确的非线性和不可预测的变化保留时间维度。
虽然有可能实现替代调整保留时间的方法,其他方法一般都需要鉴定的化合物,这并不总是可行的,或手动操作,阻止试图自动化高通量数据分析。
数据集描述
这个示例使用两个样品PAe000153和PAe000155可以从肽图谱[2]。样品都LC-ESI-MS扫描四个盐从酿酒酵母蛋白质分数包含超过1000肽。酵母样本处理不同的化学物质(甘氨酸和丝氨酸)为了得到两个生物多样性样本。时间调整这两个数据集是最具挑战性的病例之一[3]。数据集不分布与MATLAB®。必须下载数据集到一个本地目录或你自己的库。或者,你可以尝试其他的数据集在公共数据库对蛋白质数据可用,如生鱼片数据存储库。如果你收到任何错误相关的java堆内存或空间,尽可能增加java堆空间描述在这里。LC / MS数据分析需要从操作系统扩展的内存;如果你收到“记忆”错误在运行这个例子,试着增加虚拟内存(或交换空间)的操作系统或者尝试设置3 gb开关(仅32位Windows®XP),阐述了这些技术文档。
阅读和从XML文件中提取峰值的列表包含强度数据样本处理丝氨酸和甘氨酸处理样品。
ser = mzxmlread (005 _1.mzxml)(ps, ts) = mzxml2peaks (ser,“水平”1);g = mzxmlread (005 a.mzxml)(pg, tg) = mzxml2peaks (g,“水平”1);
ser =结构体字段:扫描:x1 struct [5610] mzXML: [1 x1 struct]指数:[1 x1 struct] g =结构体字段:扫描:x1 struct [5518] mzXML: [1 x1 struct]指数:[1 x1 struct]
使用MSPPRESAMPLE函数来重新取样数据集的峰的高度和位置,同时保留质量/电荷维度。数据集重新取样与5000年都有一个共同的网格质量/电荷值。比较常见的网格是最理想的可视化,为微分分析。
(mz, y) = msppresample (ps, 5000);[MZg, Yg] = msppresample (pg, 5000);
使用MSHEATMAP函数可视化两个样本。在处理热点图这是一种常见的技术显示离子强度的对数,这增强了colormap的动态范围。
fh1 = msheatmap (mz, ts,日志(y),“决议”,0.15);标题(“丝氨酸治疗”)fh2中= msheatmap (MZg、tg、日志(Yg),“决议”,0.15);标题(“甘氨酸治疗”)


详细检查偏差问题
注意您可以可视化数据集分别;然而,向量的时间有不同的大小,因此热量地图有不同的行数(或y和Yg有不同数量的列)。此外,采样率不是常数时间向量之间的转变并不是线性的。
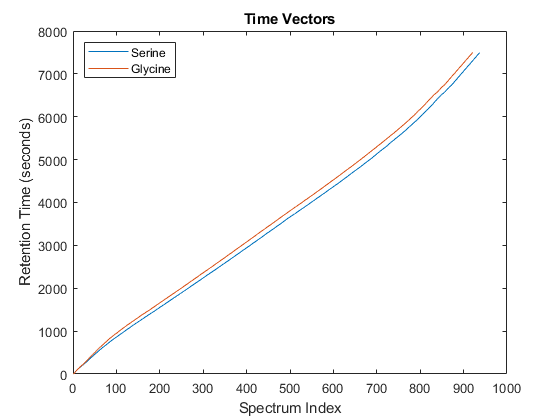
谁(“y”,“Yg”,“t”,tg的)图的阴谋(1:元素个数(ts), ts, 1:元素个数(tg), tg)传说(丝氨酸的,甘氨酸的,“位置”,“西北”)标题(“时间向量”)包含(“频谱指数”)ylabel (的保留时间(秒))
类属性名称大小字节Yg 5000 x921 18420000单y 5000 x937 18740000单tg 921 x1 7368双ts 937 x1 7496双
观察同一地区的兴趣在这两个数据集,您需要在每个矩阵计算出适当的行索引。例如,检查肽峰值在480 - 520年Da MZ范围1550 - 1900秒保留时间范围,你需要找到最接近这个范围的匹配时间向量,然后放大每个图:
ind_ser = samplealign (ts (1550; 1900));图(fh1);轴([480 520 ind_ser]) ind_gly = samplealign (tg (1550; 1900));图(fh2中);轴([480 520 ind_gly '])


即使你放大相同的范围内,你仍然可以观察到轴的右上的肽转移在保留时间维度。在样本处理丝氨酸,这个峰值的中心似乎发生在大约1595秒,而在样本处理甘氨酸假定相同肽发生在大约1630秒。这将阻止你一个准确的比较分析,即使你重新取样的数据集向量。除了保留时间的变化,数据集似乎在质量/电荷校准不当维度,因为山峰没有一个紧凑的形状在连续的光谱。
质量/电荷校准和增强的矩阵
在校正保留时间之前,您可以使用一种方法提高样本类似描述的一个例子观察和预处理用连字符连接代谢物和蛋白质/肽质谱数据集分析。为简便起见,我们只显示MATLAB代码没有任何进一步的解释:
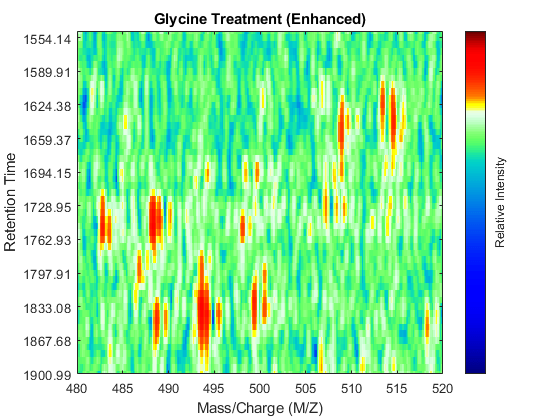
科幻小说= @ (x) 1-exp (x / 5 e7);%尺度函数DF = @ (R, S)√(科幻小说(R(:, 2))科幻(S (:, 2)))。^ 2 + (R (: 1) - s (: 1)) ^ 2);CMZ = (315: .10:680) ';%常见的细网格质量/电荷载体上%对齐的山峰丝氨酸样本MZ方向赖= 0(大小(CMZ));为i = 1:元素个数(ps)如果~ rem(我250),流(“% d…”,我);结束(k, j] = samplealign ((CMZ, LAI)双(ps{我}),“乐队”,1.5,“差距”(0 - 2),“距离”DF);赖赖= *为;赖赖(k) = (k) + p{我}(j, 2);psa{我1}= (CMZ (k) p{我}(j, 2)];结束%对齐的山峰甘氨酸样品MZ方向赖= 0(大小(CMZ));为i = 1:元素个数(pg)如果~ rem(我250),流(“% d…”,我);结束(k, j] = samplealign ((CMZ, LAI)、双(pg{我}),“乐队”,1.5,“差距”(0 - 2),“距离”DF);赖赖= *为;赖赖(k) = (k) + pg{我}(j, 2);pga{我1}= (CMZ (k) pg{我}(j, 2)];结束% Peak-preserving重新取样(mz, y) = msppresample (psa, 5000);[MZg, Yg] = msppresample (pga, 5000);%高斯滤波Gpulse = exp(闲置*(10:10)。^ 2)。/笔(exp (0。* (10:10)。^ 2));Gpulse Ysf = convn (y,“相同”);Ygf = convn (Yg、Gpulse“相同”);%的可视化fh3 = msheatmap (mz, ts,日志(Ysf),“决议”,0.15);标题(“丝氨酸治疗(增强)”轴([480 520 ind_ser ']) fh4 = msheatmap (MZg、tg、日志(Ygf),“决议”,0.15);标题(“甘氨酸治疗(增强)”轴([480 520 ind_gly '])
250年……500年……750年……250年……500年……750年……

色谱对齐
在这一点上,你有质量/电荷校准和平滑两种LC / MS数据集,但你仍无法执行一个微分分析由于数据集有一个小的偏差以及保留时间轴。
您可以使用SAMPLEALIGN正确的漂移在色谱领域。首先,你应该检查数据,寻找最坏情况下的转变,这将帮助你评估乐队约束。通过两个火地图平移可以观察到常见的肽峰值不转移超过50秒。使用输入参数SHOWCONSTRAINTS显示约束空间的时间扭曲操作和评估,如果动态规划(DP)算法可以处理这个问题的大小。在这种情况下,你有不到12000个节点。通过省略输出参数,SAMPLEALIGN只显示约束没有运行DP算法。还要注意,输入信号过滤和增强的数据集,但这些已经upsampled 5000 MZ值,如果你使用所有非常计算要求。因此,使用函数MSPALIGN获得减少的质量/电荷值列表(RMZ)表示最强烈的山峰在哪里,然后使用SAMPLEALIGN函数的指数也找到mz(或MZg)最佳匹配质量/电荷减少向量:
RMZ = mspalign ([ps; pg) ';RMZ idx = samplealign (mz,“宽度”1);%与这些输入参数%操作相当于找到%为每个RMZ在最近邻% mz。samplealign ([ts Ysf (idx:)”], [tg Ygf (idx:) '),“乐队”,50岁,“showconstraints”,真正的)


SAMPLEALIGN使用欧氏距离作为默认分数配对样本。在LC / MS数据集每个样本对应一个频谱在给定的时间,因此,每一对之间的互相关匹配光谱提供了更好的距离度量。SAMPLEALIGN允许您定义自己的度量来计算光谱之间的距离,也可以想象一个度量,奖励更多的光谱双相匹配的高离子强度的峰值而不是低离子强度噪声峰值。使用输入参数重量从输入,删除第一列代表保留时间,所以光谱之间的评分标准是只基于离子强度。
cc = @(徐,Yu)(平均(徐。* Yu, 2)。^ 2)。/意味着(徐,徐* 2)/意味着(Yu . *, 2);乌兰巴托= @ (X) bsxfun (@minus, X,意味着(X, 2));df = @ (x, y) 1-cc(乌兰巴托(x)乌兰巴托(y));(i, j) = samplealign (ts Ysf (idx:)], [tg Ygf (idx:) '),“乐队”,50岁,…“距离”df,“重量”(虚假的真实(元素个数(idx))));fh5 =图;情节(ts (i)、tg (j));网格标题(“扭曲函数”)包含(数据集的保留时间与丝氨酸)ylabel (数据集的保留时间与甘氨酸)fh6 =图;持有在情节(ts(我)+ tg (j)) / 2, ts (i) tg (j))标题(转移函数的)包含(的保留时间)ylabel (保留时间转变之间的数据集)

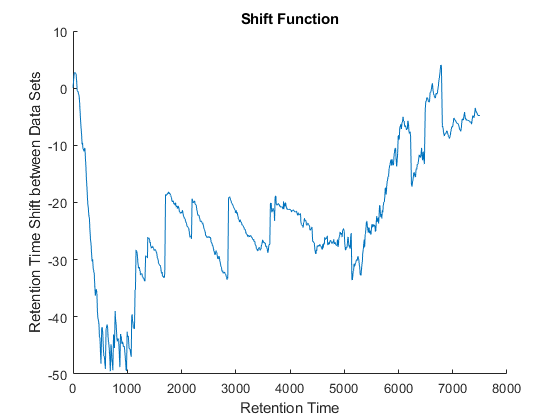
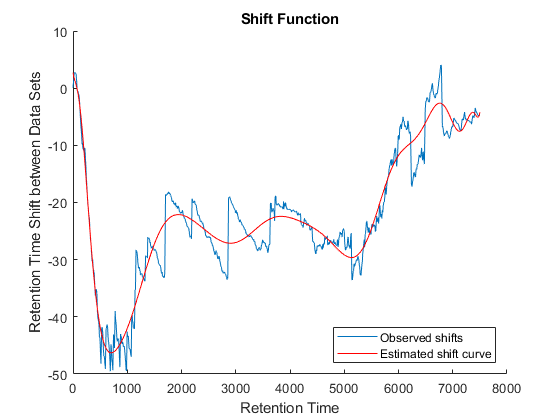
因为它预计,这两个数据集之间的真正转移函数是连续的,可以平滑的观察到的变化或回归之间的连续模型来创建一个变形模型两个时间轴。在这个例子中,我们简单地回归漂流到一个多项式函数:
(p p_structμ)= polyfit ((ts (i) + tg (j)) / 2, ts (i) tg (j), 20);科幻小说= @ (z) polyval (p (z-mu(1))。/μ(2));图(fh6)情节(tg,科幻小说(tg),“r”)传说(观察到的变化的,“估计转变曲线”,“位置”,“东南”)
比较分析
进行比较分析,重新取样LC / MS数据集向量一个共同的时间。重采样时我们使用估计功能转移到正确的保留时间维度。在这个例子中,每个谱在两个数据集是转移转移函数的距离的一半。在多个对齐的数据集的情况下,可以计算两两之间的转移函数的所有数据集,然后应用修正的一个共同的参考以这样一种方式,总体变化最小化[4]。
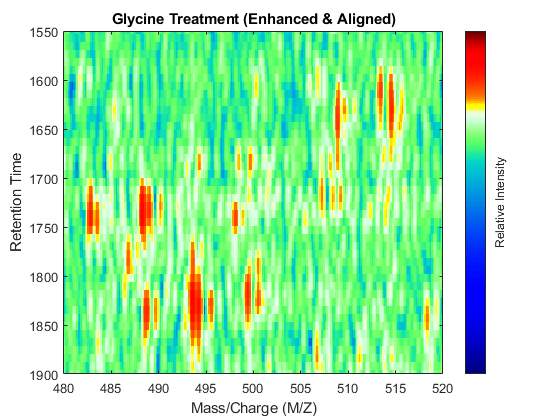
t = (max (min (tg)、min (ts)):指(diff (tg)): min (max (tg)、马克斯(ts))) ';%的可视化fh7 = msheatmap (mz t日志(interp1q (ts, Ysf’, t +科幻(t) / 2) '),“决议”,0.15);标题(“丝氨酸治疗(增强&对齐)”)fh8 = msheatmap (MZg t日志(interp1q (tg, Ygf’, t-sf (t) / 2) '),“决议”,0.15);标题(“甘氨酸治疗(增强&对齐)”)


交互地或以编程方式比较两个加强地区之间和时间对齐的数据集,您可以链接两个热图的轴。因为热量地图现在使用定期间隔时间向量,可以放大使用轴函数,而不必搜索适当的矩阵的行索引。
linkaxes (findobj ([fh7 fh8),“标签”,“MSHeatMap”)轴([480 520 1550 1900)

引用
[1]Listgarten, j .和Emili。,"Statistical and computational methods for comparative proteomic profiling using liquid chromatography tandem mass spectrometry", Molecular and Cell Proteomics, 4(4):419-34, 2005.
[2]Desiere F。,et al., "The Peptide Atlas Project", Nucleic Acids Research, 34:D655-8, 2006.
[3]王子,j.t和马克特。,"Chromatographic alignment of ESI-LC-MS proteomics data sets by ordered bijective interpolated warping", Analytical Chemistry, 78(17):6140-52, 2006.
[4]安德拉德l和Manolakos静电的,"Automatic Estimation of Mobility Shift Coefficients in DNA Chromatograms", Neural Networks for Signal Processing Proceedings, 91-100, 2003.