酵母异质丙蛋白循环中的参数扫描,参数估计和敏感性分析
此示例示出了如何建立,模拟和利用文献所取的路径在SimBiology®分析的模型。
参考
酵母异络蛋白循环的定量表征。Tau-mu yi,Hiroaki Kitano,和Melvin I. Simon。PNAS(2003)卷。100,10764-10769。
目标
创建用于酵母TMY101(重量)株的模型示出了野生型G蛋白失活的(催化)速率。
为TMY111(mut)菌株创造一个变体,其显示突变体(未催化)的G蛋白灭活率。
模拟并存储从两个模型中的数据。
比较野生型途径,突变途径和实验数据之间的G蛋白激活的时间表。
执行参数扫描以确定在感兴趣的物种上改变参数的值的效果。
使用实验数据估算模型参数值。
执行敏感性分析以确定模型参数对感兴趣的物种有哪些影响,以及影响到什么程度。
背景
在酵母中,G蛋白在交配反应中的信号转导是一个特征明确的信号转导途径。细胞分泌的信息素激活“a”细胞中的g蛋白偶联因子受体(Ste2p),从而导致各种细胞反应,包括细胞周期阻滞和新蛋白质的合成。G蛋白和G蛋白偶联受体(GPCRs)是制药行业药物发现的重点。许多已上市的药物以GPCRs为靶点——一些例子包括减少胃酸(雷尼替丁,以组胺H2受体为靶点)、偏头痛(舒马曲坦,以血清素受体亚型为靶点)、精神分裂症(奥氮平,以血清素和多巴胺受体为靶点)和过敏(地氯雷他定,以组胺受体为靶点)。此外,一些估计表明,GPCRs是40%的药物发现努力的目标重点。一种方法是建立GPCR信号通路模型,分析和预测下游效应和相关通路的效应。这个例子检验了酵母信息素反应途径中G蛋白循环的模型构建、模拟和分析。
该图是用于模拟酵母G蛋白质循环的概念框架的图形表示。
使用以下缩写:
l =配体
r =受体
gd = g alpha-gdp
Gbg=Gβγ的自由水平
ga = g alpha - gtp
G =无活性异质型G-蛋白(含有Gα和Gβ-Gamma)
null=源或接收器
Sst2为G蛋白调节因子(RGS) Sst2p

途径反应
如该图所示,该循环可以被浓缩成一组生化反应:
1) 受体-配体相互作用(可逆反应)
L + R < - > RL
2)异三聚体G蛋白的形成
Gd + Gbg - >g
3) G-蛋白活化-请注意,RL出现在方程式的两侧,因为RL是反应的修饰剂或催化剂。该反应既不产生也不消耗RL。
RL + G - > GA + GBG + RL
4) 受体合成和降解(视为可逆反应,代表降解和合成)
[R < - >空
5)受体 - 配体降解
rl - > null
6)G蛋白失活,通过Sst2p在野生型菌株TMY101催化和在突变株TMY111与所述SST2基因破坏未催化。
Ga->Gd
所有值已转化为物种量的分子,分子/秒或1 /秒用于速率参数。
构建SimBiology®模型野生型途径
创建一个名为“异三聚体G蛋白重量”一个SimBiology模型对象。
modelObj = sbiomodel('杂漆g蛋白质重量);
添加受体 - 配体相互作用(可逆反应)。
reactionObj1 = addreaction(modelObj,'l + R < - > RL',......“姓名”,“Receptor-ligand互动”);
使用“批量”动力学法进行反应。该模型是使用质量动作动力学构建的所有反应。
kineticlawObj1 = addkineticlaw(reactionObj1,“MassAction”);
添加正向和反向速率参数。
addparameter(modelobj,“kRL”3.32 e-18);addparameter(modelobj,“kRLm”, 0.01);
在动态法律对象中分配parametervariablenames。这将Parametervariables映射到动态法律对象中的ParametervariaBlenames,以便确定反应速率。
kineticlawObj1.ParameterVariableNames={“kRL”,“kRLm”};
SimBiology会自动为反应中的每个参与物种创建物种对象。设定这些物种的初始数量。
%设定初始量为“L”modelObj.Reactions (1) .Reactants(1)。InitialAmount = 6.022 e17;%设置'r'的初始金额模型对象反应(1).反应物(2).初始量=10000.0;%保留“RL”的初始值为默认值(0.0)
用于第一反应ReactionRate现已配置。
yreticobj1.realction.
ans ='krl * l * r - krlm * rl'
完成野生型模式
要创建野生型应变的模型(TMY101),请添加其余的反应和参数,为每个反应创建动力学定律对象,并为动力学法分配参数变量。
添加并配置异三聚体G蛋白形成反应。
ressobj2 = AddReacection(Modelobj,“Gd+Gbg->G”,......“姓名”,'G蛋白复杂的形成');kineticlawObj2 = addkineticlaw(reactionObj2,“MassAction”);addparameter(modelobj,'kg1', 1.0); kineticlawObj2.ParameterVariableNames='kg1';%设置“钆”初始金额modelobj.reactions(2)。重演(1).initialamount = 3000;%设定为“GBG”初始量Modelobj.reactions(2)。重演(2).initialamount = 3000;%为“G”设置初始金额模型对象反应(2).产物(1).初始量=70下载188bet金宝搏00;
添加和配置的G蛋白活化反应。
ressobj3 = addReacection(Modelobj,'G + RL - > GA + GBG + RL',......“姓名”,“G蛋白活化”);KineticLawobj3 = addkineticlaw(ressionobj3,“MassAction”);addparameter(modelobj,“克格勃”,1.0e-5);KineticLawobj3.ParametervariaBlenames =“克格勃”;%设定为“嘎”初始量模型对象反应(3).产物(1).初始量=0.下载188bet金宝搏0;
添加并配置受体合成和降解的反应。
reactionObj4=添加反应(modelObj,'r < - > null',......“姓名”,'R合成/降解'); kineticlawObj4=添加KineticLaw(反应BJ4,“MassAction”);addparameter(modelobj,'krdo', 4.0的军医);addparameter(modelobj,“kRs”, 4.0); kineticlawObj4.ParameterVariableNames={'krdo',“kRs”};
添加和配置为受体 - 配体降解反应。
reactionObj5 = addreaction (modelObj,'rl - > null',“姓名”,'rl劣化');KineticLawobj5 = Addkineticlaw(ressionobj5,“MassAction”);addparameter(modelobj,'krd1',0.0040);kineticlawObj5.ParameterVariableNames ='krd1';
添加和配置为G蛋白失活的反应。
ressobj6 = addreacrection(modelobj,'GA - >钆',“姓名”,'gprotein失活');KineticLawobj6 = Addkineticlaw(Astryovj6,“MassAction”);addparameter(modelobj,'kgd',0.11);KineticLawobj6.ParametervariaBlenames ='kgd';
检查所有反应的ReactionRate。
得到(modelobj.reaciptions,{'反应',“反应速率”})
ANS = 6×2单元阵列{ 'L + R < - > RL'} { 'KRL * L * R - kRLm * RL'} { '的Gd + GBG - > G'} { 'KG1 *的Gd * GBG'} {”G + RL - > GA + GBG + RL '} {' KGA * G * RL '} { 'R < - >空'} { 'kRdo * R - KRS'} { 'RL - >空'} {' kRD1* RL”} { '镓 - >的Gd'} { 'KGD *嘎'}
模拟野生型模型并绘制结果
为了注意到物种Ga的快速上升和随后的下降,模拟模型600秒并存储结果。
将默认配置设置对象的“停止时间”从10s (simulationTime)修改为600s。此外,不要记录配体'L' (modelObj.Species(1))的数据,因为它的值比其他物种高几个数量级。这使得物种在图中可视化更加方便。为了实现这一点,定义StatesToLog包括除'L'以外的所有物种。
configsetObj=getconfigset(modelObj);configsetObj.StopTime=600;configsetObj.SolverOptions.AbsoluteTolerance=1.e-9;configsetObj.RuntimeOptions.StatesToLog=......sbioselect (modelObj'类型',“物种”,'在哪里',“姓名”,'〜=',“L”);
模拟模型并将结果返回到三个变量“时间”,“数据”和“名称”。
[时间,数据,名称] = sbiosmulate(modelobj);
绘制数据。
图(时间,数据);传奇(名字,'地点',“东北朝”); xlabel(的时间(秒));ylabel('物种金额');网格在;

为突变菌株创建模型变体
突变株的g蛋白循环模型不同于活性g蛋白(Ga)失活的速率。该速率由速率参数kGd的值决定。您可以使用Variant对象表示突变菌株。SimBiology Variant存储SimBiology模型的一个或多个属性的替代值,例如物种的InitialAmount或参数的Value。
将名为“突变体”的变体添加到模型中。
variantObj=添加变量(modelObj,'突变');
将内容添加到所述变体,以指定的0.004为参数KGD的替代值。
addcontent(variantObj{'范围','kgd',“价值”,0.004});
模拟突变途径并绘制结果
使用突变体变体对象模拟模型。这在仿真期间将0.004的值应用于参数KGD。返回Simdata对象中的模拟结果。除了存储Simbiology仿真数据外,Simdata对象还提供了数据访问,绘图和分析的方法。
将变体对象的活动属性设置为true并模拟。
variantObj.Active=true;突变数据=sbiosimulate(modelObj);
绘制使用虚线的数据。也可以看看sbioplot.方便地绘制Simdata对象。
绘图(muntatdata.Time、muntatdata.Data、,“线型”,' - ');传说(mutantData.DataNames,'地点',“东北朝”); xlabel(的时间(秒));ylabel('物种金额');网格在;

比较野生型和突变型途径中活性G蛋白物种(Ga)的行为。
gaindex = strcmp(名称,'ga');野生型结果的%指数[tmut,xmut]=按名称选择(修改数据,'ga');绘图(时间,数据(:,gaindex),tmut,xmut,' - '); xlabel(的时间(秒));ylabel('物种金额'); 传奇({'Ga(wt)','Ga(突变)'},'地点',“东北朝”);网格在;

执行参数扫描
G蛋白失活的速率低得多相对于野生型(KGD = 0.004 VS KGD = 0.11),它比在上面的对比观察时间解释了更高水平的活性的G蛋白(Ga)的的突变株中.在KGD的变化如何影响的Ga水平的更详细的研究,执行多个仿真,其中KGD的值被改变在一定范围的值中的参数扫描。下面的例子说明过的KGD五个值的参数扫描;增加迭代次数,更改参数的值Linspace.下面的功能。
生成五个均匀分布的kGd值,范围从0.001到0.15。
kgdvalues = linspace(1e-3,0.15,5);
将参数扫描结果存储在SimData对象数组中。初始化变量以保存此数组。
Scandata = [];
为加速仿真准备模型。
sbioaccelerate(modelobj);
循环kGdValues并对每个值执行模拟。使用模型上的突变变量来修改仿真过程中使用的kGd值。
对于kgd = kgdvalues.%,在变体中设置的KGD所需的值。variantObj.Content{1}{4}=kGd;%模拟模型,将结果存储在SimData对象中。sd=sbiosimulate(modelObj);斯堪的纳维亚语=[斯堪的纳维亚语;sd];结尾
斯堪的数据现在是Simdata对象的五个元素数组。每个对象都包含从参数扫描中运行的数据。
从SIMDATA对象阵列和情节上的单轴提取嘎timecourses。下面的代码构造的情节一步工序;另外,看sbioplot.和sbiosubplot.
[tscan,xscan] = selectbyname(scandata,'ga');跳频=图;抓住在;对于c = 1:5 plot(tscan {c},xscan {c});str = sprintf(' k = %5.3f',kgd值(c));文本(tscan{c}(end)、xscan{c}(end)、str);结尾%注释情节。轴(gca (fh),'正方形');标题('改变KGD的价值:对Ga'的影响'); xlabel(的时间(秒));ylabel('物种金额');网格在;抓住离开;

参数估计 - 背景
在对生物系统进行建模时,通常需要包括数值未知或仅大致已知的参数。如果系统中有一个或多个物种的实验数据可用,则可以通过改变这些参数并寻找能够使模型模拟结果与实验数据最佳拟合的值来估计这些参数的值。
在该示例的本部分中,我们在尝试将G蛋白模型适合实验数据的上下文中探索参数估计功能。
参数估计-比较模型结果与实验数据
在实验数据方面,参考文献图5中包含了G蛋白活性组分的时间历程。
存储实验时间和状态数据。
texpt = [0 10 30 60 110 210 300 450 600]'GAFRACEXPT = [0 0.35 0.4 0.36 0.39 0.39 0.24 0.17 0.2]';data = groupeddata(表(texpt,gafracexpt));data.properties.independentvariablename ='tExpt';
而不是将该实验数据转换为绝对量的GA,而是使用非常量参数和重复分配规则将此分数添加到模型。
gafracobj = modelobj.addparameter('GAFRAC','constantvalue',0);gafracrule = modelobj.addrule('GAFRAC = GA /(GA + G + GD)','repeatedAssignment')
GAFRACRULE = SIMBIOGUCALY RULE ARRAY索引:RULETYPE:规则:1 Repectedassignment GAFRAC = GA /(GA + G + GD)
变化对配置集中RuntimeOptions登录GaFrac。
configsetObj.RuntimeOptions.StatesToLog = GaFracObj;
取消激活突变体变量。
variantobj.active = false;
模拟模型,将结果存储在Simdata对象中。
sdWild = sbiosimulate(modelObj);
获取“GAFRAC”的数据以稍后在绘图中使用。
[otdld,gafracwild] = selectbyname(sdwild,'GAFRAC');
重新采样的模拟结果到实验时间矢量。
sdwildresampled =重组(sdwild,texpt,'pchip');
获得重采样数据供种“嘎”。
[~,GaFracWildResampled]=selectbyname(sdWildResampled,'GAFRAC');
计算R平方值测量所述模拟和实验数据之间的配合。
SST =常规(GAFRACEXPT - 平均值(GAFRACEXPT))^ 2;SSE =常规(GAFRACEXPT - GAFRACWILDRES采样)^ 2;RSQUARE = 1-SSE / SST;
将模拟结果绘制于GA的实验数据。
跳频=图;情节(tExpt GaFracExpt,“罗”); 传奇文本={'实验'}; 头衔('适合GAFRAC'的实验数据); xlabel(的时间(秒));ylabel(“物种数量”);抓住在;情节(tWild GaFracWild);LegendText {END + 1} = SPRINTF('原始,R ^ 2 =%4.2f', rSquare);传奇(legendText {:});网格在;

参数估计 - 估计单个模型参数
从参数扫描中,我们已经看到参数kGd的值对物种Ga的时间进程有显著影响。让我们看看是否可以通过改变kGd的值来改善模型结果与实验数据的拟合。
对实验数据执行参数估计,优化KGD的值。绘图有关迭代的信息,同时优化进展,最多15个迭代。
paramToEst = estimatedInfo('kgd');kgdobj = sbioselect(modelobj,“姓名”,'kgd');选择= OptimSet('plotfcns',@ OptimplotFval,'maxiter'15);结果1 = sbiofit(modelobj,数据,'GaFrac = GaFracExpt',paramtoest,......[],'fminsearch',选择);estvalues1 =结果1.Parameterestimates.
估计StandardError estValues1 = 1 x3表名称 _______ ________ _____________ {' kGd} 0.12142 - 0.0018554
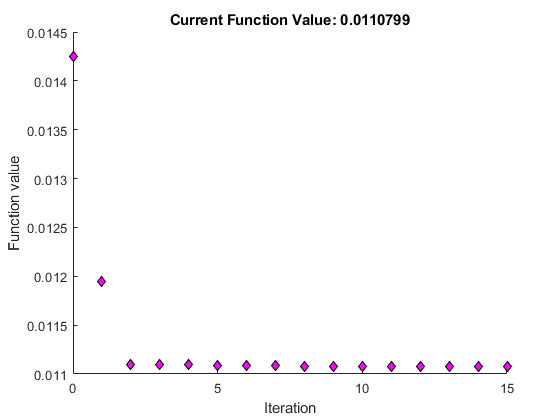
将KGD的估计值存储在新模型变体中。
OptimVariantobj = addVariant(Modelobj,'优化KGD'); addcontent(optimVariantObj{'范围','kgd',“价值”,estValues1.Estimate});
激活新变体并使“突变”变体失活。
OptimVariantobj.active = True;mutantvariantobj = getvariant(modelobj,'突变');mutantVariantObj.Active = FALSE;
使用KGD的估计值模拟模型。
sdEst1 = sbiosimulate(modelObj);
绘制GAFRAC的数据,并与上一个结果进行比较。
[T1,GaFracEst1] = selectbyname(sdEst1,'GAFRAC');SDEST1RESAMPLED =重组(SDEST1,TEXPT,'pchip');[〜,GaFracEst1Resampled] = selectbyname(sdEst1Resampled,'GAFRAC'); sse1=范数(GaFracExpt-GaFracEst1Resampled)^2;rSquare1=1-sse1/sst;数字(fh);图(t1,GaFracEst1,'m-');LegendText {END + 1} = SPRINTF('KGD更改,R ^ 2 =%4.2f',rsquare1);传奇(legendText {:});
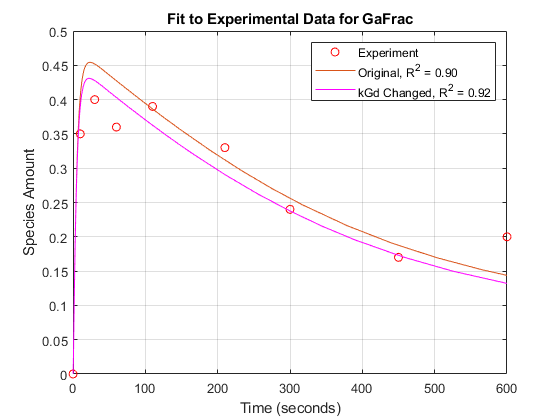
从R-square值可以看出,新的kGd估价值与实验数据的拟合略好一些。如果kGd的原始值只是一个粗略的估计,我们可以将这些结果解释为对原始估计的确认或对其的改进。
敏感性分析 - 背景
到目前为止,我们一直对活性G蛋白的动态行为,物种Ga。参数扫描显示,该物种受到治疗G蛋白灭活的速率常数KGD的值的显着影响。使用参数估计,我们发现通过优化KGD的价值,我们能够更好地适合GA的实验时间库。
要问的自然问题是,模型的其他参数会影响GA水平,这些效果的大小是什么?灵敏度分析允许您通过计算相对于型号参数值或物种初始条件(“输入因子”)计算一个或多个物种(“输出”)的时间相关的衍生物来回答这些问题。
敏感性分析 - 计算敏感性
在模型中计算Ga的灵敏度。完全归一化灵敏度,以便它们可以相互比较。
停用模型上的突变变量对象,以便使用kGd在其原始值计算灵敏度。
OptimVariantobj.active = false;
设置在模型configset的灵敏度计算。
%在求解器选项中打开SensitiveSanalysis。configsetObj.SolverOptions.SensitivityAnalysis=true;%为灵敏度分析配置灵敏度输出和输入。sensitivityOpt = configsetObj.SensitivityAnalysisOptions;GaObj = sbioselect(modelObj,'类型',“物种”,“姓名”,'ga');sensitivityopt.outputs = gaobj;params = sbioselect(modelobj,'类型','范围','在哪里',“姓名”,'〜=','GAFRAC');sensitivityOpt。输入=参数;sensitivityOpt。归一化='满的';
模拟模型。
sdsens = sbiosmulate(modelobj);
从SimData对象中提取灵敏度数据,并绘制计算出的灵敏度。
[t,r,sensointpuls,sensinputs] = getsensmatrix(sdsens);r =挤压(r);数字;情节(t,r);标题(“GA关于各种参数的归一化敏感性”); xlabel(的时间(秒));ylabel('灵敏度');传奇(SensInputs,'地点',“东北朝”);网格在;

参数估计-估计多个参数
结果表明,Ga不仅对参数kGd敏感,而且对kGa、kRs和kRD1也敏感。(在情节中,其他敏感因素几乎是零。)改变这四个参数可以使其更符合实验数据。
估计这四个参数以匹配目标数据。使用以前配置的优化选项和模型中的当前参数值作为优化的起点。
选择参数kGa、kRs、kRD1和kGd进行估算。
paramsToEst = estimatedInfo ({“克格勃”,“kRs”,'krd1','kgd'});
如果在configset启用参数估计会忽略了敏感性分析选项。在求解器选项,以避免警告关闭SensitivityAnalysis。
configsetobj.solveroptions.sensitivityAnalysis = false;结果2. sbiofit(modelobj,数据,'GaFrac = GaFracExpt',paramsToEst,......[],'fminsearch',选择);estValues2 = result2.ParameterEstimates
Estvalues2 = 4x3表名估计标准误差____________________________________'} 4.0309C-06 3.0309E-06 {'KRD1'} 0.0031018 0.0031018 0.003778

将四个参数的估计值存储在新模型变体中。
OptimVariantobj2 = addVariant(Modelobj,“四参数优化”); addcontent(optimVariantObj2{'范围',“克格勃”,“价值”估计值(1)});addcontent(optimVariantObj2{'范围',“kRs”,“价值”估计(2)});addcontent(optimVariantObj2{'范围','krd1',“价值”estValues2.Estimate (3)});addcontent (optimVariantObj2, {'范围','kgd',“价值”,estvalues2.imate(4)});
现在模拟与新参数估计值模型。
OptimVariantobj.active = false;optimVariantObj2。积极= true;sdEst2 = sbiosimulate (modelObj);
与以前的结果进行比较。
[T2,GaFracEst2] = selectbyname(sdEst2,'GAFRAC');SDEST2RESAMPLED =重组(SDEST2,TEXPT,'pchip');[~, GaFracEst2Resampled] = selectbyname(sdEst2Resampled,'GAFRAC'); sse2=标准(GaFracExpt-GAFRACEST2采样)^2;rSquare2=1-sse2/sst;数字(fh);图(t2,GaFracEst2,'G-');LegendText {END + 1} = SPRINTF('4常数发生变化,R ^ 2 =%4.2f',rsquare2);传奇(legendText {:});
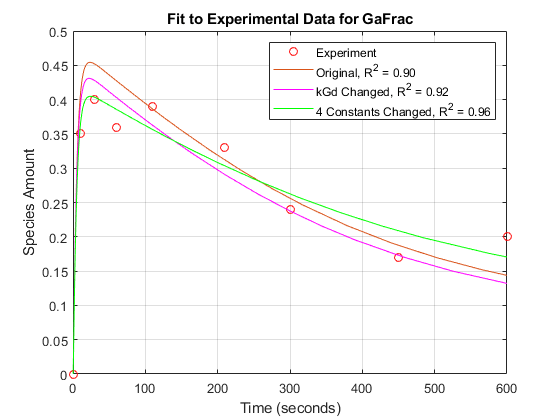
通过参数估计可自由地改变四个参数,适合于实验数据进一步提高。显示的优化迭代显示目标函数降低,R范围值增加。
需要注意的是四参数估计在这里进行的可能或不可能是生物学相关的,是仅供参考。
还要注意的是,将估计结果存储在变量中可以方便地在模拟模型的不同版本之间来回切换。目前有四种版本:原始版本、突变版本和基于参数估计结果的两种版本。
结论
该示例介绍了使用G蛋白信号模型进行模型建筑,模拟和分析的偶像功能的各个方面。
您还可以从以下列表中选择一个网站: