利用神经网络预测蛋白质二级结构
这个例子展示了一个二级结构预测方法,该方法使用前馈神经网络和深度学习工具箱™提供的功能。
这是一个简单的例子,旨在说明建立神经网络的步骤,以预测蛋白质的二级结构。它的配置和训练方法并不一定是解决当前问题的最佳方案。
介绍
神经网络模型试图模拟大脑中发生的信息处理过程,并被广泛应用于各种应用,包括自动模式识别。
Rost-Sander数据集[1]由结构跨越相对广泛的结构域类型、组成和长度的蛋白质组成。该文件RostSanderDataset.mat包含该数据集的子集,其中每个残基的结构分配被报告为每个蛋白质序列。
负载RostSanderDataset.matN =元素个数(allSeq);id = allSeq(7)。头%对给定蛋白质序列的注释seq = int2aa (allSeq (7) . sequence)%的蛋白质序列str = allSeq(7)。结构%结构赋值
id = '1CSE-ICOMPLEX(SERINEPROTEINASE-INHIBITOR)03-JU' seq = 'KSFPEVVGKTVDQAREYFTLHYPQYNVYFLPEGSPVTLDLRYNRVRVFYNPGTNVVNHVPHVG' str = ' ccchhhcccchhhhhhhhhcccceeeeecccccceeeeeecccccceeeeeecccccceecccceec '
在本例中,您将根据训练阶段观察到的结构模式,构建一个神经网络来学习给定蛋白质中每个残基的结构状态(螺旋、薄片或线圈)。由于以下方法中某些步骤的随机性,每次训练网络或模拟预测时,数值结果可能略有不同。为了确保结果的重现性,我们将全局随机生成器重置为加载文件中包含的已保存状态,如下所示:
rng (savedState);
定义网络架构
对于当前的问题,我们定义了一个具有一个输入层、一个隐藏层和一个输出层的神经网络。输入层在每个输入氨基酸序列中编码一个滑动窗口,并对窗口中中心残基的结构状态进行预测。我们根据给定残差位置的二级结构与预测点[2]两侧的8个残差之间的统计相关性,选择大小为17的窗口。每个窗口位置使用大小为20的二进制数组进行编码,每个氨基酸类型都有一个元素。在每组20个输入中,给定位置的氨基酸类型对应的元素设置为1,而所有其他输入设置为0。因此,输入层由R = 17x20的输入单元组成,即17组,每组20个输入。
在下面的代码中,我们首先通过创建汉克尔矩阵,为每个蛋白质序列确定与大小为W的滑动窗口相对应的所有可能的子序列,其中第i列表示从原始序列的第i个位置开始的子序列。然后,对于窗口中的每个位置,我们创建一个大小为20的数组,如果给定位置的剩余数的数字表示为j,则将第j个元素设置为1。
W = 17;滑动窗口大小%% ===输入二值化为i = 1:N seq = double(allSeq(i).Sequence);%当前序列赢得=汉克尔(seq (1: W), seq (W:结束);%所有可能的滑动窗口项目= 0 (20 * W,大小(赢,2));%输入矩阵为当前序列为k = 1:尺寸(赢,2)指数= 20 * (0:w1) +赢得(:,k);%为每个位置k输入数组项目(指数(k) = 1;结束allSeq(我)。P =项目;结束
我们神经网络的输出层由三个单元组成,每个单元对应于所考虑的结构状态(或类),它们使用二进制编码。为了建立神经网络的目标矩阵,我们首先从数据中获得与滑动窗口相对应的所有可能子序列的结构分配。然后我们考虑每个窗口的中心位置,并使用以下二进制编码转换相应的结构赋值:1 0 0表示线圈,0 1 0表示薄片,0 0 1表示螺旋。
cr =装天花板(W / 2);中心剩余位置%% ===目标二值化为i = 1:N str = double(allSeq(i).Structure);当前结构转让赢得=汉克尔(str (1: W), str (W:结束);%所有可能的滑动窗口myt = false(3,尺寸(胜利,2));myt(1,:) = win(cr,:) == double(“C”);myT(2,:) = win(cr,:) == double(“E”);myT(3,:) = win(cr,:) == double(“H”);allSeq(我)。T =我;结束
你可以通过执行以下等价代码,以一种更简洁的方式来执行上述两个步骤中描述的输入和目标矩阵的二值化:
% ===输入和目标的简洁二值化为i = 1:N seq = double(allSeq(i).Sequence);赢得=汉克尔(seq (1: W), seq (W:结束);%并发输入(滑动窗口)% ===输入矩阵二值化allSeq(我)。P= kron(win,ones(20,1)) == kron(ones(size(win)),(1:20)');% ===目标矩阵二值化allSeq(我)。T= allSeq(i).Structure(repmat((W+1)/2:end-(W-1)/2,3,1)) ==...repmat ((“甚至”), 1,长度(allSeq(我).Structure) - w + 1);结束
一旦我们为每个序列定义了输入矩阵和目标矩阵,我们就创建一个输入矩阵,P,和目标矩阵,T,表示输入网络的所有序列的编码。
% ===构造输入和目标矩阵P =双([allSeq.P]);%的输入矩阵T =双([allSeq.T]);%的目标矩阵
创建神经网络
二级结构预测问题可以看作是一个模式识别问题,训练网络识别当观察到给定滑动窗口中的特定残差时最可能出现的中心残差的结构状态。我们使用上面定义的输入和目标矩阵创建一个模式识别神经网络,并指定一个大小为3的隐藏层。
hsize = 3;网= patternnet (hsize);net.layers {1}%隐藏层net.layers {2}%输出层
ans = Neural Network Layer name: 'Hidden' dimensions: 3 distanceFcn: (none) distanceParam: (none) distance: [] initFcn: 'initnw' netInputFcn: 'netsum' netInputParam: (none) positions: [] range: [3x2 double] size: 3 topologyFcn: (none) transferFcn: 'tansig' transferParam: (none) userdata: (your custom info) ans = Neural Network Layer name:'Output' dimensions: 0 distanceFcn: (none) distanceParam: (none) distance: [] initFcn: 'initnw' netInputFcn: 'netsum' netInputParam: (none) positions: [] range: [] size: 0 topologyFcn: (none) transferFcn: 'softmax' transferParam: (none) userdata: (your custom info)
神经网络的训练
模式识别网络使用默认的缩放共轭梯度算法进行训练,但也可以使用其他算法(参见深度学习工具箱文档获取可用函数列表)。在每个训练周期,训练序列通过上面定义的滑动窗口一次一个残差呈现给网络。每个隐藏单元使用传递函数对从输入层接收的信号进行变换logsig产生在0或1之间的输出信号,模拟神经元的射击[2]。调整权重,使得来自每个单元的观察到的输出和目标矩阵指定的所需输出之间的误差最小化。
% ===使用log sigmoid作为传递函数net.layers{1}。transferFcn =“logsig”;% ===训练网络(净,tr) =火车(净、P、T);
在培训期间,将打开培训工具窗口并显示进度。训练细节,如算法,性能标准,考虑的错误类型等。

使用功能视图生成神经网络的图形视图。
查看(网)

神经网络训练过程中出现的一个常见问题是数据过拟合,网络倾向于记住训练示例,而不学习如何对新情况进行概括。改进泛化的默认方法称为早期停止,它包括将可用的训练数据集划分为三个子集:(i)训练集,用于计算梯度和更新网络权值和偏差;(ii)验证集,在训练过程中监测其误差,因为当数据过度拟合时,其误差往往会增加;(iii)测试集,其误差可用于评估数据集划分的质量。
在使用函数时火车,默认情况下,对数据进行随机划分,将60%的样本分配给训练集,20%分配给验证集,20%分配给测试集,但可以通过指定属性应用其他类型的划分net.divideFnc(默认dividerand).从下面的调查中可以看出,这三个子集中的残留物的结构组成具有可比性:
(i, j) =找到(T (:, tr.trainInd));= sum(i == 1)/length(i);Etrain = sum(i == 2)/length(i);Htrain = sum(i == 3)/length(i);(i, j) =找到(T (:, tr.valInd));Cval = sum(i == 1)/length(i);Eval = sum(i == 2)/length(i);Hval = sum(i == 3)/length(i);(i, j) =找到(T (:, tr.testInd));Ctest = sum(i == 1)/length(i); Etest = sum(i == 2)/length(i); Htest = sum(i == 3)/length(i); figure() pie([Ctrain; Etrain; Htrain]); title(“训练数据集的结构分配”);传奇(“C”,“E”,“H”图()派()[Cval;Eval;Hval]);标题(“验证数据集中的结构赋值”);传奇(“C”,“E”,“H”图()派()[ct;浓度梯度法;ht]);标题(“测试数据集的结构分配”);传奇(“C”,“E”,“H”)



这个函数plotperform在训练迭代过程中显示训练、验证和测试错误的趋势。
图()plotperform (tr)

当出现以下情况之一时,训练过程停止(见net.trainParam)是满足。例如,在考虑的训练中,当验证错误增加到指定的迭代次数(6)或达到允许的最大迭代次数(1000)时,训练过程停止。
% ===显示训练参数net.trainParam% ===地块验证检查和梯度图()plottrainstate (tr)
ans = Training Window Feedback showWindow: true Show CommandLine Feedback showCommandLine: false CommandLine Frequency Show: 25 Maximum Epochs Epochs: 1000 Maximum Training Time Time Time: Inf Performance Goal Goal: 0 Minimum Gradient min_grad: 1e-06 Maximum Validation Checks max_fail: 6 Sigma Sigma:5e-05 Lambda

网络响应分析
为了分析网络响应,我们考虑训练网络的输出,并将其与预期结果(目标)进行比较,来检验混淆矩阵。
O = sim(净,P);图()plotconfusion (T O);

对角线单元格显示了每个结构类正确分类的剩余位置的数量。非对角线细胞显示了错误分类的残留位置的数量(例如,螺旋位置被预测为盘绕位置)。对角线的格对应正确分类的观察结果。每个单元格中都显示了观测次数和观测总数的百分比。图中最右边的一列显示了预测属于正确和错误分类的每个类别的所有示例的百分比。这些指标通常分别称为精度(或阳性预测值)和错误发现率。图底部的一行显示了属于正确和错误分类的每个类的所有示例的百分比。这些指标通常分别称为召回率(或真阳性率)和假阴性率。图右下角的单元格显示了整体的准确性。
我们还可以考虑受试者工作特征(ROC)曲线,即真实阳性率(敏感性)与假阳性率(1 -特异性)的曲线。
图()plotroc (T O);

改进神经网络以获得更准确的结果
我们定义的神经网络相对简单。为了提高预测精度,我们可以尝试以下方法之一:
增加训练向量的数量。增加专门用于训练的序列的数量需要一个更大的蛋白质结构的策划数据库,具有适当的螺旋、螺旋和薄片元素的分布。
增加输入值的数量。增加窗口大小或添加更多相关信息,如氨基酸的生化特性,都是有效的选择。
使用不同的训练算法。不同的算法在内存和速度要求上有所不同。例如,缩放共轭梯度算法相对较慢,但内存效率高,而Levenberg-Marquardt算法更快,但在内存方面要求更高。
增加隐藏神经元的数量。通过添加更多隐藏单元,我们通常能够获得具有更好性能的更复杂网络,但我们必须小心不要过度拟合数据。
我们可以在创建模式识别网络时指定更多的隐藏层或增加隐藏层的大小,如下图所示:
Hsize = [3 4 2];net3 = patternnet (hsize);hsize = 20;net20 = patternnet (hsize);
我们还可以将网络初始权值分配给[2]中报告的研究建议的-0.1到0.1范围内的随机值,通过设置net20。信息战和net20.lw.属性如下:
% ===在-范围内分配随机值。1和。1的权重net20.iw {1} = -.1 +(.1 + .1)。* rand(size20.iw {1});net20.lw {2} = -.1 +(.1 + .1)。* rand(大小(net20.lw {2}));
一般来说,较大的网络(包含20个或更多的隐藏单元)在蛋白质训练集上获得更好的精度,但在预测精度上的精度较差。因为一个20个隐藏单元的网络包含近7000个权值和偏差,所以网络通常能够紧密地拟合训练集,但失去了泛化的能力。强化训练和预测精度之间的折衷是神经网络的基本限制之一。
net20 =火车(net20、P、T);O20 = sim (net20 P);numWeightsAndBiases =长度(getx (net20))
numWeightsAndBiases = 6883
通过单击训练工具窗口中相应的按钮,可以显示训练、验证和测试子集的混淆矩阵。
评估网络性能
您可以通过计算预测质量指数[3]来详细评估结构预测,[3]表明对特定状态的预测有多好,以及是否发生了过度预测或过低预测。我们定义索引pcObs (S)状态S (S = {C、E、H})残留物的数量准确的预测在国家年代,除以残留物的数量在国家美国类似的,我们定义索引pcPred (S)国家年代残留物的数量正确预测在国家年代,除以残留物的数量预计在国家年代。
(i, j) =找到(专业(O));[u, v] =找到(T);% ===计算在观察给定状态时正确预测的分数pcObs(1) = sum(i == 1 & u == 1)/sum (u == 1);% C状态pcObs(2) = sum(i == 2 & u == 2)/sum (u == 2);国家E %pcObs(3) = sum(i == 3 & u == 3)/sum (u == 3);% H状态% ===计算在预测给定状态时正确预测的百分比pcPred(1) = sum(i == 1 & u == 1)/sum (i == 1);% C状态pcPred(2) = sum(i == 2 & u == 2)/sum (i == 2);国家E %pcPred(3) = sum(i == 3 & u == 3)/sum (i == 3);% H状态% ===比较预测的质量指标图()栏([PCOBS'PCPRED'] * 100);ylabel(“正确预测的位置(%)”);甘氨胆酸ax =;斧子。XTickLabel = {“C”;“E”;“H”};传奇({“观察”,“预测”});
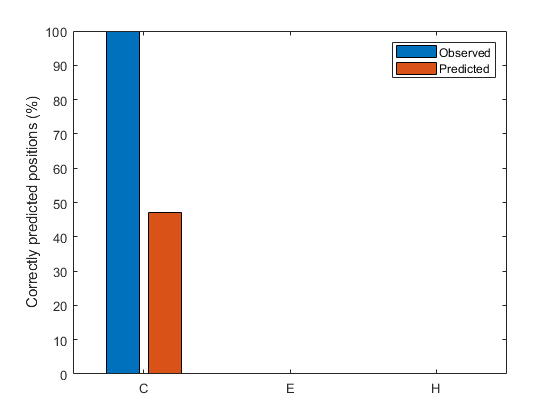
这些质量指标对预测精度的解释是有用的。事实上,在预测技术倾向于过度预测/低估给定状态的情况下,高/低的预测精度可能只是人为因素,并不能为技术本身提供质量度量。
结论
本文提出的方法是根据邻近蛋白质残基的结构状态来预测其结构状态。然而,在预测蛋白质中结构元素的含量时,还有进一步的限制,如每个结构元素的最小长度。具体来说,一个螺旋被分配到任何一组四个或更多连续的残基,而一个表被分配到任何一组两个或更多连续的残基。为了整合这类信息,可以创建一个额外的网络,以便第一个网络从氨基酸序列预测结构状态,第二个网络从结构状态预测结构元素。
nntraintool (“关闭”)%关闭神经网络训练工具
参考
[1] Rost .,和Sander .,“预测蛋白质二级结构的准确率高于70%”,《分子生物学杂志》,32(2):584- 599,1993。
[2]“基于神经网络的蛋白质二级结构预测”,中国生物医学工程学报,21(1):1 - 7,1989。
[3] kabch, W., and Sander, C.,“预测蛋白质二级结构有多好?”,FEBS Letters, 155(2):179- 82,1983。
你也可以从以下列表中选择一个网站: