nbintest
小样本量计数资料的非配对假设检验
描述
测试= nbintest (X,Y)
短读计数采用负二项分布建模。
每一行数据的方差和平均值通过所有行上的回归函数相连。
X和Y必须有相同的行数和至少2列,但不一定相同的列数。行X和Y对应变量、特征或基因,如不同基因的基因表达量。列通常是时间点或病人。
测试是一个NegativeBinomialTest对象中存储的双面p值pValue财产。
当您想用小样本量(最多10个数量级)对短读计数数据(来自高通量分析,如RNA-Seq或ChIP-Seq)执行非配对假设检验时,请使用此功能。例如,使用这个函数来判断在给定的基因中,两种条件下读取计数的观察差异是否显著。
测试= nbintest (X,Y,名称,值)名称,值对参数。
请注意
的诊断图NegativeBinomialTest返回的对象nbintest在解释p值之前。这些图允许您查看模型假设是否正确,以及所使用的方差链接是否适合于数据。
例子
对短读计数数据进行非配对假设检验
这个例子展示了如何对来自两种不同生物条件的合成短读计数数据执行非配对假设检验。
本例中的数据包含5000个基因的合成基因计数数据,代表两种不同的生物条件,如患病细胞和正常细胞。对于每种情况,有5个样本。只有10%的基因(500个基因)有差异表达。具体来说,其中一半(250个基因)恰好过表达3倍。其他250个基因是3倍的低表达。其余的基因表达数据来自两种情况下相同的负二项分布。每个样本也有一个不同的大小因子(即覆盖范围或采样深度)。
加载数据。
负载(“nbintest_data.mat”,“K”,“H0”);
的变量K包含基因计数数据。行表示基因,列表示样本。在本例中,前五列表示来自第一个条件的示例。其他五列表示来自第二个条件的样本。的前几行K.
: K (1:5)
ans =5×1013683 14140 8281 14309 12208 8045 9446 11317 14597 14592 16028 16805 9813 16476 9901 10927 13348 16999 17036 814 492 910 758 521 7353 870 936 15870 16453 9857 16454 14267 9671 10971 13624 17151 17205 9422 9393 5798 8174 5381 6315 7752 9869 9795
在这个例子中,当基因没有差异表达时,零假设是正确的。的变量H0包含布尔指标,表明哪些基因的零假设为真(标记为1)。换句话说,H0包含已知的标签,您稍后将使用这些标签与预测结果进行比较。
总和(H0)
ans = 4500
在5000个基因中,有4500个基因在这个合成数据中没有差异表达。
对来自两种情况的样本进行非配对假设检验nbintest.假设数据来自一个负二项分布,其中方差通过在[1]中描述的局部回归均值平滑函数与均值相连“VarianceLink”来“LocalRegression”.
tLocal = nbintest (K(: 1:5)、K (:, 6:10)“VarianceLink”,“LocalRegression”);
使用plotVarianceLink绘制每个实验条件(X和Y条件)的散点图,并使用公共尺度上的样本方差与条件相关均值的估计值进行比较。对两个轴都使用线性比例。包括曲线的所有其他连接选项设置“比较”来真正的.
plotVarianceLink (tLocal“规模”,“线性”,“比较”,真正的)
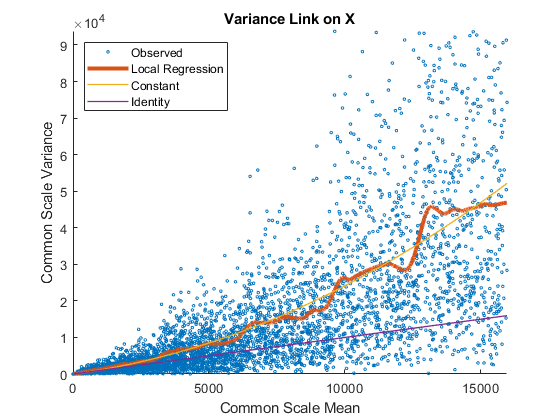
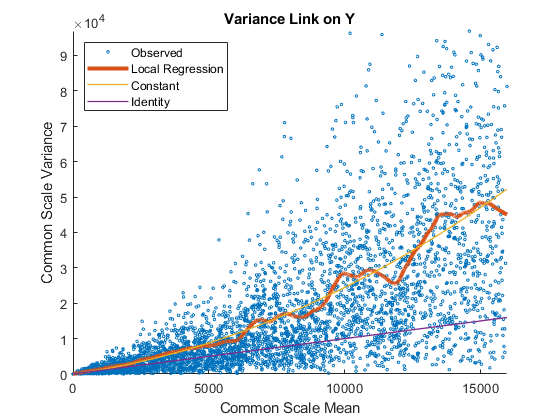
的身份直线表示泊松模型,其中方差等于[3]中描述的均值。观察数据似乎是过度分散的(即,大多数点都高于身份线)。的常数Line表示负二项模型,其中方差是散粒噪声项(均值)和一个常数乘以均值的平方,如[2]所述。的当地的回归和常数链接选项似乎更适合过于分散的数据。
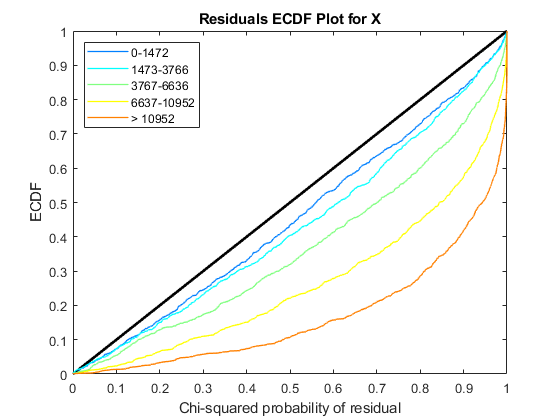
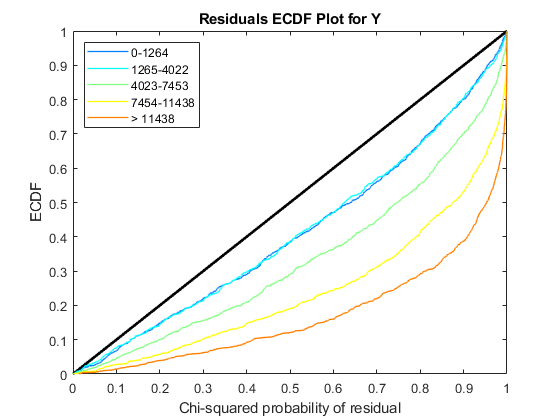
使用plotChiSquaredFit评估方差回归的拟合优度。它绘制了卡方概率的经验CDF (ecdf)。概率是观察到的和估计的方差之间的比率,由短读计数水平分层到五个等大小的箱子。
plotChiSquaredFit (tLocal)
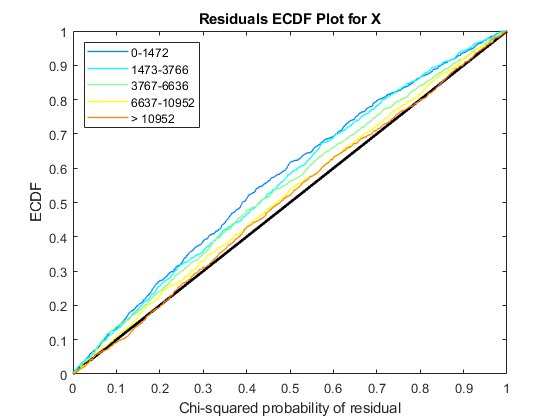
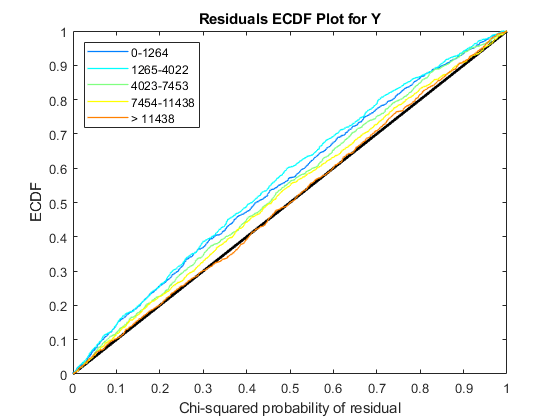
每个图显示了五条ecdf曲线。每条曲线代表五个短读计数级别中的一个。例如,蓝色的线表示在0到1264之间的低短读计数的ecdf曲线。红线代表高计数(超过11438)。
解释曲线的一种方法是检查ecdf曲线是否在对角线之上。如果它们高于这条线,则方差被高估了。如果它们低于这条线,那么方差被低估了。在这两个图中,对于较高的计数,方差似乎是正确估计的(也就是说,红线跟随对角线),但对于较低的计数水平,方差估计略高。
为了评估假设检验的表现,使用已知的标签和预测的p值构造一个混淆矩阵。
confusionmat (H0 (tLocal。pValue >措施)
ans =2×2493 7 5 4495
在500个差异表达基因中,493个被正确预测(真阳性),其中7个被错误预测为无差异表达基因(假阴性)。在4500个没有差异表达的基因中,4495个被正确预测(真阴性),其中5个被错误预测为差异表达基因(假阳性)。
为了进行比较,再次进行假设检验,假设计数由泊松分布建模,其中方差与均值相同。
tPoisson = nbintest (K(: 1:5)、K (:, 6:10)“VarianceLink”,“身份”);
绘制ecdf曲线。注意所有的曲线都在对角线以下,这意味着方差被低估了。因此,负二项模型对数据拟合较好。
plotChiSquaredFit (tPoisson)
输入参数
输出参数
参考文献
[1] Anders, S.和Huber, W.(2010)。序列计数数据的差异表达分析。基因组生物学、11 (10):R106。
[2] Robinson, m.d.和Smyth, G.K.(2008)。负二项离散度的小样本估计,及其在SAGE数据中的应用。生物统计学,9:321 - 332。
[3] Marioni, j.c., Mason, c.e., Mane, s.m., Stephens, M.,和Gilad, Y.(2008)。RNA-seq:技术重现性的评估和与基因表达阵列的比较。基因组研究,16:1509 - 1517。
你也可以从以下列表中选择一个网站: